
29 Mar
2010
29 Mar
'10
4:46 p.m.
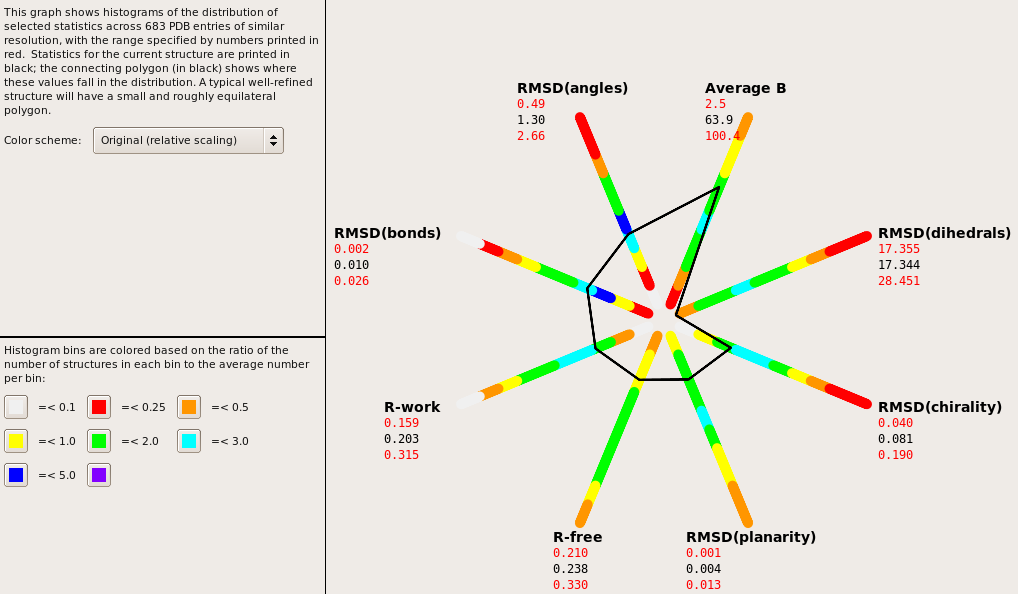
Hi all, I'm working on a ~3 angstrom structure of a protein with approximately 800 residues. The structure is mostly helical with interspersed turns. I've been using Phenix exclusively for the refinements and have a model that I believe to be pretty good with the exception of the dihedrals (see attached). I've looked at and employed some of the general suggestions for low resolution structures here: http://strucbio.biologie.uni-konstanz.de/ccp4wiki/index.php/Refinement#Refin... I tried using "discard_psi_phi=False" but it only resulted in a change dihedrals of +0.10 with minimal changes to Ramachandran outliers. I'm unsure how to proceed and would love to hear any thoughts/opinions/suggestions. Thanks for all the help! -Jon
{kind=link}