staraniso/phenix.refine

Dear all, I am trying to validate and then (hopefully) deposit a structure generated using the autoproc/staraniso staraniso_alldata-unique.mtz file as input for phenix.refine. Autoproc also produces a cif file ( Data_1_autoPROC_STARANISO_all.cif) specifically for deposition. Long story short, the PDB validation server complains about the lack of a freeR set for both files. I realized that, at least for the cif file, the r_free_flag is missing (but why does the .mtz for the isotropic dataset work??),so I then tried to use for validation the *.reflections.cif file that can be generated by phenix.refine. This file can actually produce a validation report, but I still have some questions: 1) Is it proper to use the .reflections.cif file for this purpose? During the upload I do see some errors (see pics); also, the final results show various RSRZ outliers in regions of the structure that look reasonably good by looking at the maps on coot, which seems odd ... 2) In case the *.reflections.cif is not adequate/sufficient for deposition (I sent an inquiry to the PDB, but they did not respond yet), can I just add a _refln.status column to the autoproc cif file (within the loop containing the r_free_flag) where I insert “f” for r_free_flag = 0 and “o” everywhere else? Thank you in advance, Andrea
{kind=link}

Dear Andrea I also experienced that the PDB cannot appreciate the Data_1_autoPROC_STARANISO_all.cif file due to the lack of Rfree set. This would be solvable, like in Refmac5 (CCP4i2), if you can upload second reflection file that contains the free R flags used for refinement? - However, CCP41/Refmac5 does not (yet) read .cif reflection files. As far as I know, Phenix Refine does not (yet) neither. - But Refmac5 does merge two .mtz reflection files into a sinlle hklout.mtz that you can find under job1 directory. This is what I currently use to deposit to the PDB. - I understand .cif files (both model and reflections) have the advantage that they are editable. Perhaps there is a good reflection.cif file editor around? - A solution would be to have the option during PDB deposition, of adding the reflection file used for refinement and thus containing the R free set, that are then automatically recognized as reflection files and merged (like for hklout.mtz in Refmac where this option was probably incorporated for re-refinement with re-processed data). Problem today is that only one reflection file is permitted and you would indeed not get the correct validation report outputted. In other words, one data file from crystal data, and another one for crystal refinement and including map coefficients, or like we do it in the lab, would allow compatibility with different ways of re-refinement and perhaps the independence from the EDS server (that failed on me last week, meaning there are no electron densities in the validation files and meanwhile the submission is "locked" and also no answer from the PDB staff yet, so what do we do: we submit the poor validation file to the journal and be more subject to rejection by reviewers. What I prefer, seen the current situation, is to send reviewers the model coordinates and map coefficients). all the very best Julie On 17/12/22 02:32, Andrea Piserchio wrote:
Dear all,
I am trying to validate and then (hopefully) deposit a structure generated using the autoproc/staraniso staraniso_alldata-unique.mtz file as input for phenix.refine.
Autoproc also produces a cif file ( Data_1_autoPROC_STARANISO_all.cif) specifically for deposition.
Long story short, the PDB validation server complains about the lack of a freeR set for both files. I realized that, at least for the cif file, the r_free_flag is missing (but why does the .mtz for the isotropic dataset work??),so I then tried to use for validation the *.reflections.cif file that can be generated by phenix.refine. This file can actually produce a validation report, but I still have some questions:
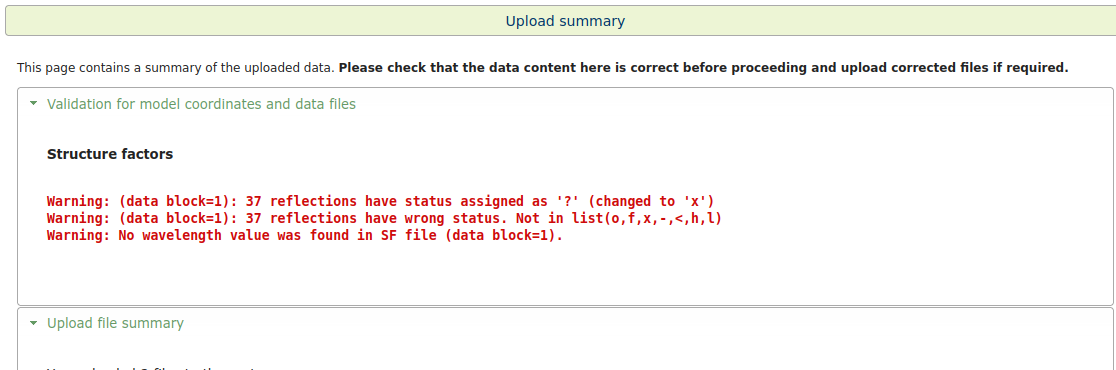
1) Is it proper to use the .reflections.cif file for this purpose? During the upload I do see some errors (see pics); also, the final results show various RSRZ outliers in regions of the structure that look reasonably good by looking at the maps on coot, which seems odd ...
2) In case the *.reflections.cif is not adequate/sufficient for deposition (I sent an inquiry to the PDB, but they did not respond yet), can I just add a _refln.status column to the autoproc cif file (within the loop containing the r_free_flag) where I insert “f” for r_free_flag = 0 and “o” everywhere else?
Thank you in advance,
Andrea
_______________________________________________ phenixbb mailing list [email protected] http://phenix-online.org/mailman/listinfo/phenixbb Unsubscribe: [email protected] -- _______________ Julie Bouckaert , chercheur CNRS UMR 8576 à l’Université de Lille http://ugsf-umr-glycobiologie.univ-lille1.fr/spip.php?article217&lang=fr Unité de Glycobiologie Structurale et Fonctionnelle (UGSF) IRI, Office 131, 50 Avenue de Halley, 59658 Villeneuve d’Ascq, France Tél. +33 (0) 362 53 17 29 Mobile +32 (0) 498 57 14 65

Dear Andrea and Julie,
We also encountered the same problem recently, and Clemens and Gerard from Global Phasing (cc:ed) were extremely helpful by quickly adding to the latest (Nov 21 2022) snapshot of BUSTER (https://www.globalphasing.com/buster/) a version of aB_deposition_combine that can correctly merge an autoPROC/STARANISO reflection file with the results of either PHENIX or REFMAC refinement. I personally haven’t tried the latter, but the combination autoPROC/STARANISO + PHENIX mmCIF definitely works (and PDB validation/submission did not complain!).
Have a look here:
http://www.globalphasing.com/buster/ReleaseNotes/ReleaseNotes-BUSTER_snapsho...
Clemens and Gerard, perhaps a note about this could also be added to the autoPROC/STARANISO documentation (if it’s not already there)? Although aB_deposition_combine “belongs” to BUSTER, users may not go search for it there - as opposed to the STARANISO documentation - if they decided to go the PHENIX way…
HTH,
Luca
--
Luca Jovine, Ph.D.
Professor of Structural Biology, Member of EMBO and the Nobel Assembly
Karolinska Institutet
Department of Biosciences and Nutrition
Medicinaren 25 Neo
Blickagången 16, SE-141 83 Huddinge, Sweden
E-mail: [email protected]
W3: http://jovinelab.org
From:

Hi, two hopefully relevant points: - phenix.refine always produces an MTZ file that contains the copy of all inputs plus all is needed to run refinement (free-r flags, for example). So if you use that file for deposition you have all you need. - Unless there are strongly advocated reasons to do otherwise in your particular case, you better use in refinement and deposit the original data and NOT the one touched by any of available these days magic sticks (that "correct" for anisotropy, sharpen or else!). Other comments:
- However, CCP41/Refmac5 does not (yet) read .cif reflection files. As far as I know, Phenix Refine does not (yet) neither.
Phenix supports complete input / outputs of mmcif/cif format. For example, phenix.refine can read/write model and reflection data in cif format. It's been this way for a long time now. Pavel On 12/16/22 17:32, Andrea Piserchio wrote:
Dear all,
I am trying to validate and then (hopefully) deposit a structure generated using the autoproc/staraniso staraniso_alldata-unique.mtz file as input for phenix.refine.
Autoproc also produces a cif file ( Data_1_autoPROC_STARANISO_all.cif) specifically for deposition.
Long story short, the PDB validation server complains about the lack of a freeR set for both files. I realized that, at least for the cif file, the r_free_flag is missing (but why does the .mtz for the isotropic dataset work??),so I then tried to use for validation the *.reflections.cif file that can be generated by phenix.refine. This file can actually produce a validation report, but I still have some questions:
1) Is it proper to use the .reflections.cif file for this purpose? During the upload I do see some errors (see pics); also, the final results show various RSRZ outliers in regions of the structure that look reasonably good by looking at the maps on coot, which seems odd ...
2) In case the *.reflections.cif is not adequate/sufficient for deposition (I sent an inquiry to the PDB, but they did not respond yet), can I just add a _refln.status column to the autoproc cif file (within the loop containing the r_free_flag) where I insert “f” for r_free_flag = 0 and “o” everywhere else?
Thank you in advance,
Andrea
_______________________________________________ phenixbb mailing list [email protected] http://phenix-online.org/mailman/listinfo/phenixbb Unsubscribe: [email protected]
So,
Both the phenix-generated mtz file (silly me for not checking this first)
and the cif file generated by aB_deposition_combine can be uploaded on the
PDB server.
Thank you all for your help!!
Andrea
On Sat, Dec 17, 2022 at 5:06 PM Pavel Afonine
Hi,
two hopefully relevant points:
- phenix.refine always produces an MTZ file that contains the copy of all inputs plus all is needed to run refinement (free-r flags, for example). So if you use that file for deposition you have all you need.
- Unless there are strongly advocated reasons to do otherwise in your particular case, you better use in refinement and deposit the original data and NOT the one touched by any of available these days magic sticks (that "correct" for anisotropy, sharpen or else!).
Other comments:
- However, CCP41/Refmac5 does not (yet) read .cif reflection files. As far as I know, Phenix Refine does not (yet) neither.
Phenix supports complete input / outputs of mmcif/cif format. For example, phenix.refine can read/write model and reflection data in cif format. It's been this way for a long time now.
Pavel
On 12/16/22 17:32, Andrea Piserchio wrote:
Dear all,
I am trying to validate and then (hopefully) deposit a structure generated using the autoproc/staraniso staraniso_alldata-unique.mtz file as input for phenix.refine.
Autoproc also produces a cif file ( Data_1_autoPROC_STARANISO_all.cif) specifically for deposition.
Long story short, the PDB validation server complains about the lack of a freeR set for both files. I realized that, at least for the cif file, the r_free_flag is missing (but why does the .mtz for the isotropic dataset work??),so I then tried to use for validation the *.reflections.cif file that can be generated by phenix.refine. This file can actually produce a validation report, but I still have some questions:
1) Is it proper to use the .reflections.cif file for this purpose? During the upload I do see some errors (see pics); also, the final results show various RSRZ outliers in regions of the structure that look reasonably good by looking at the maps on coot, which seems odd ...
2) In case the *.reflections.cif is not adequate/sufficient for deposition (I sent an inquiry to the PDB, but they did not respond yet), can I just add a _refln.status column to the autoproc cif file (within the loop containing the r_free_flag) where I insert “f” for r_free_flag = 0 and “o” everywhere else?
Thank you in advance,
Andrea
_______________________________________________ phenixbb mailing list [email protected] http://phenix-online.org/mailman/listinfo/phenixbb Unsubscribe: [email protected]

Dear Pavel, I thought I ought to respond to the second paragraph of your message, as I find it rather disappointing that you chose to deal with this matter in such a dismissive and unhelpful manner. When users decide that they are willing to go to the trouble of trying to deposit the data produced by STARANISO, it is presumably because they derived some benefit from using it as input into building and/or refining their model. Clemens has "walked the extra mile" to enable the deposition of such data along with refinement results obtained not only with BUSTER but also with REFMAC and phenix.refine, out of a desire to enable users to do what they want to do, rather than try and dissuade them from it. You deride "available these days magic sticks (that "correct" for anisotropy, sharpen or else!)". The UCLA Diffraction Anisotropy Server has been doing exactly that for over a decade and a half without incurring this kind of sarcasm. As for objecting to corrections, sharpening and truncation, should that extend to the upscaling of higher-resolution data by image-wise B-factors and to the rejection of outliers, all of which take place in every single scaling and merging job? Probably not - so this remark sounds more as if it is just gratuitously picking on STARANISO rather than offering helpful advice to users. With best wishes, Gerard. -- On Sat, Dec 17, 2022 at 02:06:26PM -0800, Pavel Afonine wrote:
Hi,
two hopefully relevant points:
- phenix.refine always produces an MTZ file that contains the copy of all inputs plus all is needed to run refinement (free-r flags, for example). So if you use that file for deposition you have all you need.
- Unless there are strongly advocated reasons to do otherwise in your particular case, you better use in refinement and deposit the original data and NOT the one touched by any of available these days magic sticks (that "correct" for anisotropy, sharpen or else!).
Other comments:
- However, CCP41/Refmac5 does not (yet) read .cif reflection files. As far as I know, Phenix Refine does not (yet) neither.
Phenix supports complete input / outputs of mmcif/cif format. For example, phenix.refine can read/write model and reflection data in cif format. It's been this way for a long time now.
Pavel
On 12/16/22 17:32, Andrea Piserchio wrote:
Dear all,
I am trying to validate and then (hopefully) deposit a structure generated using the autoproc/staraniso staraniso_alldata-unique.mtz file as input for phenix.refine.
Autoproc also produces a cif file ( Data_1_autoPROC_STARANISO_all.cif) specifically for deposition.
Long story short, the PDB validation server complains about the lack of a freeR set for both files. I realized that, at least for the cif file, the r_free_flag is missing (but why does the .mtz for the isotropic dataset work??),so I then tried to use for validation the *.reflections.cif file that can be generated by phenix.refine. This file can actually produce a validation report, but I still have some questions:
1) Is it proper to use the .reflections.cif file for this purpose? During the upload I do see some errors (see pics); also, the final results show various RSRZ outliers in regions of the structure that look reasonably good by looking at the maps on coot, which seems odd ...
2) In case the *.reflections.cif is not adequate/sufficient for deposition (I sent an inquiry to the PDB, but they did not respond yet), can I just add a _refln.status column to the autoproc cif file (within the loop containing the r_free_flag) where I insert “f” for r_free_flag = 0 and “o” everywhere else?
Thank you in advance,
Andrea
_______________________________________________ phenixbb mailing list [email protected] http://phenix-online.org/mailman/listinfo/phenixbb Unsubscribe: [email protected]
phenixbb mailing list [email protected] http://phenix-online.org/mailman/listinfo/phenixbb Unsubscribe: [email protected]
Dear Gerard, I didn't mean to sound unhelpful and if I did I apologize for that! What I really meant is this.. I see the growing trend of using various data alternation techniques and then not depositing the original data but the data changed by these techniques. Clearly if this trend exists these techniques must be helpful! However, my concern is that by not depositing the original data we methods and software developers loose access to these precious original data that we could use to further improve the methods. I witnessed multiple instances of this happening with UCLA Anisotropy Server, for example, or even before that X-plor program allowing some venue to sharpen Fobs and then making it easy to continue using the sharpened version (that got me at some point!). Or even before that the standard practice of cutting off the low-resolution end of data set at about 6...8A and worse -- in the absence of appropriate bulk solvent models that was actually a good thing to do at that time! Or truncating the data by "sigma" (those classic 2-3 sigma cutoffs!) -- that was also a good thing to do when refinement programs could only use LS target functions. And now look in the PDB and see how many data sets lack those low-res or weak reflections! So, no, I am not picking on a particular tool or method, but I'm picking on the trend that makes it potentially easy to permanently loose the original data, one way or another, even with a good intention in mind. So my real message was and is: any one is of course free to use any tools available, but please deposit the original data (along with any data used to obtain the final atomic model). Perhaps someone uses these data and comes up with even better tools in the future? All the best! Pavel On 12/21/22 08:46, Gerard Bricogne wrote:
Dear Pavel,
I thought I ought to respond to the second paragraph of your message, as I find it rather disappointing that you chose to deal with this matter in such a dismissive and unhelpful manner.
When users decide that they are willing to go to the trouble of trying to deposit the data produced by STARANISO, it is presumably because they derived some benefit from using it as input into building and/or refining their model. Clemens has "walked the extra mile" to enable the deposition of such data along with refinement results obtained not only with BUSTER but also with REFMAC and phenix.refine, out of a desire to enable users to do what they want to do, rather than try and dissuade them from it.
You deride "available these days magic sticks (that "correct" for anisotropy, sharpen or else!)". The UCLA Diffraction Anisotropy Server has been doing exactly that for over a decade and a half without incurring this kind of sarcasm. As for objecting to corrections, sharpening and truncation, should that extend to the upscaling of higher-resolution data by image-wise B-factors and to the rejection of outliers, all of which take place in every single scaling and merging job? Probably not - so this remark sounds more as if it is just gratuitously picking on STARANISO rather than offering helpful advice to users.
With best wishes,
Gerard.
-- On Sat, Dec 17, 2022 at 02:06:26PM -0800, Pavel Afonine wrote:
Hi,
two hopefully relevant points:
- phenix.refine always produces an MTZ file that contains the copy of all inputs plus all is needed to run refinement (free-r flags, for example). So if you use that file for deposition you have all you need.
- Unless there are strongly advocated reasons to do otherwise in your particular case, you better use in refinement and deposit the original data and NOT the one touched by any of available these days magic sticks (that "correct" for anisotropy, sharpen or else!).
Other comments:
- However, CCP41/Refmac5 does not (yet) read .cif reflection files. As far as I know, Phenix Refine does not (yet) neither. Phenix supports complete input / outputs of mmcif/cif format. For example, phenix.refine can read/write model and reflection data in cif format. It's been this way for a long time now.
Pavel
On 12/16/22 17:32, Andrea Piserchio wrote:
Dear all,
I am trying to validate and then (hopefully) deposit a structure generated using the autoproc/staraniso staraniso_alldata-unique.mtz file as input for phenix.refine.
Autoproc also produces a cif file ( Data_1_autoPROC_STARANISO_all.cif) specifically for deposition.
Long story short, the PDB validation server complains about the lack of a freeR set for both files. I realized that, at least for the cif file, the r_free_flag is missing (but why does the .mtz for the isotropic dataset work??),so I then tried to use for validation the *.reflections.cif file that can be generated by phenix.refine. This file can actually produce a validation report, but I still have some questions:
1) Is it proper to use the .reflections.cif file for this purpose? During the upload I do see some errors (see pics); also, the final results show various RSRZ outliers in regions of the structure that look reasonably good by looking at the maps on coot, which seems odd ...
2) In case the *.reflections.cif is not adequate/sufficient for deposition (I sent an inquiry to the PDB, but they did not respond yet), can I just add a _refln.status column to the autoproc cif file (within the loop containing the r_free_flag) where I insert “f” for r_free_flag = 0 and “o” everywhere else?
Thank you in advance,
Andrea
_______________________________________________ phenixbb mailing list [email protected] http://phenix-online.org/mailman/listinfo/phenixbb Unsubscribe: [email protected]
phenixbb mailing list [email protected] http://phenix-online.org/mailman/listinfo/phenixbb Unsubscribe: [email protected]
Dear Pavel, Thank you for your most courteous and constructive reply to my message that, I must admit, had a few sharp edges. I guess I over-reacted when I came across the expression "available these days" in your mention of "magic sticks", thinking that you were referring to a recent newcomer among this category of programs - i.e. to STARANISO - while absolving the exact similar sins of the UCLA server ... ;-) . This misunderstanding and associated sensitivities now being behind us, I am in complete agreement with what you wrote to clarify and argue your own position. It is undoubtedly true that many PDB entries contain too little experimental data, and that a more proactive involvement of the PDB itself in formulating guidelines and requirements would have been helpful. Users have been left for too long without a clear picture of the fact that they should deposit at least two kinds of corroborating data along with their models, namely (1) the map coefficients used to compute the map(s) whose interpretation led to the model being deposited, as this is the most direct experiment-linked evidence for that model, and (2) the primary diffraction data themselves that led, along with the model (or even independently from it, in the case of experimental phasing), to those map coefficients. As there can be diverse notions of what is "primary" in this context - given that the most obvious meaning (i.e. the raw diffraction images) has remained out of the realm of possibilites for PDB archiving - there has been a lack of clear guidance or enforcement as to what actual experimental data should be deposited. As the STARANISO-treated diffraction data have in many cases provided maps that played a significant role in pinning down certain key components of final models, it may well have often seemed like killing two birds with one stone to deposit these data alone as the corroborating experimental information, introducing a break in the link with raw data if a description of the nature of that treatment is not included. Our reaction to this situation has been to contribute to the creation of a Subgroup on Data Collection and Processing of the PDBx/mmCIF Working Group of the wwPDB (quite a mouthful ... ) and to its activities, first towards extending the mmCIF dictionary to make clearly delineated room for jointly depositing data before and after treatment with STARANISO (or any other programs with a similar purpose) together with a description of that treatment, resulting on March 31st 2021 in the following announcement: https://www.wwpdb.org/news/news?year=2021#60638da1931d5660393084c3 . This extension also tidied up some loose ends towards better supporting the deposition of unmerged data, with the meaning of that expression at the time. Since then, a second wave of activity of the Subgroup has been aiming at further extending the mmCIF dictionary so as to accommodate unmerged data from serial diffraction experiments, as well as a more richly annotated form of unmerged data from rotation experiments. Given the strength of the concerns you express for the proper archiving in the PDB of diffraction data as close as possible to the raw measurements, I hope that you will be lending your support to this initiative, verbally first but also by contributing to the implementation work on the Phenix side that is required to make all this fly. You might even consider joining that Subgroup, so that you can be in the inner loop of the discussions and of the coordination of efforts. See you in Zoom meetings in the New Year, then ? With best wishes, Gerard. -- On Wed, Dec 21, 2022 at 02:53:43PM -0800, Pavel Afonine wrote:
Dear Gerard,
I didn't mean to sound unhelpful and if I did I apologize for that!
What I really meant is this.. I see the growing trend of using various data alternation techniques and then not depositing the original data but the data changed by these techniques. Clearly if this trend exists these techniques must be helpful! However, my concern is that by not depositing the original data we methods and software developers loose access to these precious original data that we could use to further improve the methods. I witnessed multiple instances of this happening with UCLA Anisotropy Server, for example, or even before that X-plor program allowing some venue to sharpen Fobs and then making it easy to continue using the sharpened version (that got me at some point!). Or even before that the standard practice of cutting off the low-resolution end of data set at about 6...8A and worse -- in the absence of appropriate bulk solvent models that was actually a good thing to do at that time! Or truncating the data by "sigma" (those classic 2-3 sigma cutoffs!) -- that was also a good thing to do when refinement programs could only use LS target functions. And now look in the PDB and see how many data sets lack those low-res or weak reflections! So, no, I am not picking on a particular tool or method, but I'm picking on the trend that makes it potentially easy to permanently loose the original data, one way or another, even with a good intention in mind.
So my real message was and is: any one is of course free to use any tools available, but please deposit the original data (along with any data used to obtain the final atomic model). Perhaps someone uses these data and comes up with even better tools in the future?
All the best! Pavel
On 12/21/22 08:46, Gerard Bricogne wrote:
Dear Pavel,
I thought I ought to respond to the second paragraph of your message, as I find it rather disappointing that you chose to deal with this matter in such a dismissive and unhelpful manner.
When users decide that they are willing to go to the trouble of trying to deposit the data produced by STARANISO, it is presumably because they derived some benefit from using it as input into building and/or refining their model. Clemens has "walked the extra mile" to enable the deposition of such data along with refinement results obtained not only with BUSTER but also with REFMAC and phenix.refine, out of a desire to enable users to do what they want to do, rather than try and dissuade them from it.
You deride "available these days magic sticks (that "correct" for anisotropy, sharpen or else!)". The UCLA Diffraction Anisotropy Server has been doing exactly that for over a decade and a half without incurring this kind of sarcasm. As for objecting to corrections, sharpening and truncation, should that extend to the upscaling of higher-resolution data by image-wise B-factors and to the rejection of outliers, all of which take place in every single scaling and merging job? Probably not - so this remark sounds more as if it is just gratuitously picking on STARANISO rather than offering helpful advice to users.
With best wishes,
Gerard.
-- On Sat, Dec 17, 2022 at 02:06:26PM -0800, Pavel Afonine wrote:
Hi,
two hopefully relevant points:
- phenix.refine always produces an MTZ file that contains the copy of all inputs plus all is needed to run refinement (free-r flags, for example). So if you use that file for deposition you have all you need.
- Unless there are strongly advocated reasons to do otherwise in your particular case, you better use in refinement and deposit the original data and NOT the one touched by any of available these days magic sticks (that "correct" for anisotropy, sharpen or else!).
Other comments:
- However, CCP41/Refmac5 does not (yet) read .cif reflection files. As far as I know, Phenix Refine does not (yet) neither. Phenix supports complete input / outputs of mmcif/cif format. For example, phenix.refine can read/write model and reflection data in cif format. It's been this way for a long time now.
Pavel
On 12/16/22 17:32, Andrea Piserchio wrote:
Dear all,
I am trying to validate and then (hopefully) deposit a structure generated using the autoproc/staraniso staraniso_alldata-unique.mtz file as input for phenix.refine.
Autoproc also produces a cif file ( Data_1_autoPROC_STARANISO_all.cif) specifically for deposition.
Long story short, the PDB validation server complains about the lack of a freeR set for both files. I realized that, at least for the cif file, the r_free_flag is missing (but why does the .mtz for the isotropic dataset work??),so I then tried to use for validation the *.reflections.cif file that can be generated by phenix.refine. This file can actually produce a validation report, but I still have some questions:
1) Is it proper to use the .reflections.cif file for this purpose? During the upload I do see some errors (see pics); also, the final results show various RSRZ outliers in regions of the structure that look reasonably good by looking at the maps on coot, which seems odd ...
2) In case the *.reflections.cif is not adequate/sufficient for deposition (I sent an inquiry to the PDB, but they did not respond yet), can I just add a _refln.status column to the autoproc cif file (within the loop containing the r_free_flag) where I insert “f” for r_free_flag = 0 and “o” everywhere else?
Thank you in advance,
Andrea
_______________________________________________ phenixbb mailing list [email protected] http://phenix-online.org/mailman/listinfo/phenixbb Unsubscribe: [email protected]
phenixbb mailing list [email protected] http://phenix-online.org/mailman/listinfo/phenixbb Unsubscribe: [email protected]
participants (5)
-
 Andrea Piserchio
Andrea Piserchio -
 Gerard Bricogne
Gerard Bricogne -
 julie
julie -
 Luca Jovine
Luca Jovine -
 Pavel Afonine
Pavel Afonine