modelling into positive densities

Hello, I am refining a structure solved to 1.5A by MR. Rw/Rf were 0.17/0.22 which seem acceptable to me. At the very beginning part of the protein the electron density is a bit wobbly. I am able to build the residues into the positive densities. But after phenix.refine the chain always shifts away a bit and leaves the green blobs there. (Photo: https://drive.google.com/open?id=1UngAJuEUt1S0LwPybMJLw2E1xA4cNM3R) I am thinking if this can be solved by adjusting the target weights. Or can I apply certain restraints only to those few residues? I refined XYZ (reciprocal space), XYZ (real space), individual B-factors, TLS and occupancies. Thanks in advance. Regards Sam

Hi,
Here are some suggestions:
1) For the offending residues, check the TLS groups. Maybe you can exclude
these residues from the TLS groups (using isotropic Bs for these residues
only) or come up with a better assignment.
2) From your description, it sounds like you refined XYZ, B, TLS and
occupancies right after MR. You could refine XYZ and B first, adding TLS
and occupancies later.
3) 1.5 Å resolution is typically the "border" for using individual
anisotropic ADP's. So you might want to try using anisotropic ADPs instead
of TLS. There is no guarantee that it'll work, but why not give it a try.
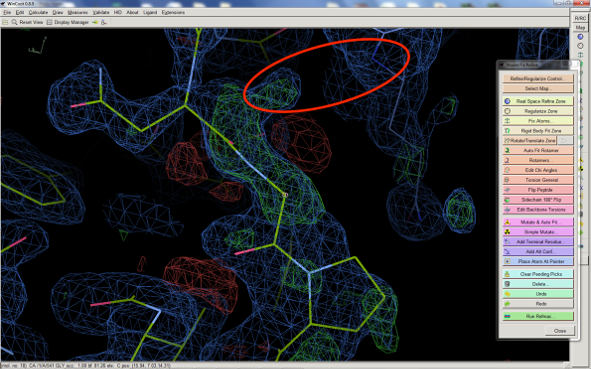
4) Unrelated to B or TLS: Check the distance between the oxygen of the
peptide unit (which is pushed out of density) and the nitrogen from the
symmetry related molecule. I circled the interaction in red in your figure.
The distance should be around 2.7-3.5 Å. I cannot tell from looking at the
screenshot. If it is shorter, nonbonded restraints could push the atoms
apart.
Best wishes,
Dorothee
[image: bolb.jpg]
On Fri, Apr 26, 2019 at 5:41 AM Sam Tang
Hello,
I am refining a structure solved to 1.5A by MR. Rw/Rf were 0.17/0.22 which seem acceptable to me. At the very beginning part of the protein the electron density is a bit wobbly. I am able to build the residues into the positive densities. But after phenix.refine the chain always shifts away a bit and leaves the green blobs there.
(Photo: https://drive.google.com/open?id=1UngAJuEUt1S0LwPybMJLw2E1xA4cNM3R )
I am thinking if this can be solved by adjusting the target weights. Or can I apply certain restraints only to those few residues?
I refined XYZ (reciprocal space), XYZ (real space), individual B-factors, TLS and occupancies.
Thanks in advance.
Regards
Sam _______________________________________________ phenixbb mailing list [email protected] http://phenix-online.org/mailman/listinfo/phenixbb Unsubscribe: [email protected]
-- Project Scientist, Molecular Biophysics and Integrated Bioimaging Lawrence Berkeley National Laboratory 1 Cyclotron Road, M/S 33R0345 Berkeley, CA 94720 Tel: (510) 486-5709 Fax: (510) 486-5909 Web: https://phenix-online.org
{kind=link}

Dear Sam, at 1.5A you might expect to see holes in the aromatic rings and at least a den in the proline residue. The region of your screen shot is quite noisy - either you integrated your data too far into the noise, or there is more than one conformation of the side chain. You can split the range of offending residues, including 1-2 either side, and see if coot models the second one reasonably. Best, Tim ________________________________ From: [email protected] [[email protected]] on behalf of Sam Tang [[email protected]] Sent: Friday, April 26, 2019 2:38 PM To: PHENIX user mailing list Subject: [phenixbb] modelling into positive densities Hello, I am refining a structure solved to 1.5A by MR. Rw/Rf were 0.17/0.22 which seem acceptable to me. At the very beginning part of the protein the electron density is a bit wobbly. I am able to build the residues into the positive densities. But after phenix.refine the chain always shifts away a bit and leaves the green blobs there. (Photo: https://drive.google.com/open?id=1UngAJuEUt1S0LwPybMJLw2E1xA4cNM3R) I am thinking if this can be solved by adjusting the target weights. Or can I apply certain restraints only to those few residues? I refined XYZ (reciprocal space), XYZ (real space), individual B-factors, TLS and occupancies. Thanks in advance. Regards Sam
Dear all
Thanks for all the advice and suggestions.
After multiple attempts I believe we were actually building too many
residues to the N-terminus (!). It also appeared the region is complicated
by extra density (seems to be EDO, likely degraded from PEG), which were
mis-treated as protein. Deleting the nearby mis-placed residues allowed the
green blobs to be fitted.
Thanks again!
Sam
On Mon, 29 Apr 2019 at 20:08, Grüne Tim (PSI)
Dear Sam,
at 1.5A you might expect to see holes in the aromatic rings and at least a den in the proline residue. The region of your screen shot is quite noisy - either you integrated your data too far into the noise, or there is more than one conformation of the side chain. You can split the range of offending residues, including 1-2 either side, and see if coot models the second one reasonably.
Best, Tim ------------------------------ *From:* [email protected] [ [email protected]] on behalf of Sam Tang [ [email protected]] *Sent:* Friday, April 26, 2019 2:38 PM *To:* PHENIX user mailing list *Subject:* [phenixbb] modelling into positive densities
Hello,
I am refining a structure solved to 1.5A by MR. Rw/Rf were 0.17/0.22 which seem acceptable to me. At the very beginning part of the protein the electron density is a bit wobbly. I am able to build the residues into the positive densities. But after phenix.refine the chain always shifts away a bit and leaves the green blobs there.
(Photo: https://drive.google.com/open?id=1UngAJuEUt1S0LwPybMJLw2E1xA4cNM3R )
I am thinking if this can be solved by adjusting the target weights. Or can I apply certain restraints only to those few residues?
I refined XYZ (reciprocal space), XYZ (real space), individual B-factors, TLS and occupancies.
Thanks in advance.
Regards
Sam
participants (3)
-
 Dorothee Liebschner
Dorothee Liebschner -
 Grüne Tim (PSI)
Grüne Tim (PSI) -
 Sam Tang
Sam Tang