
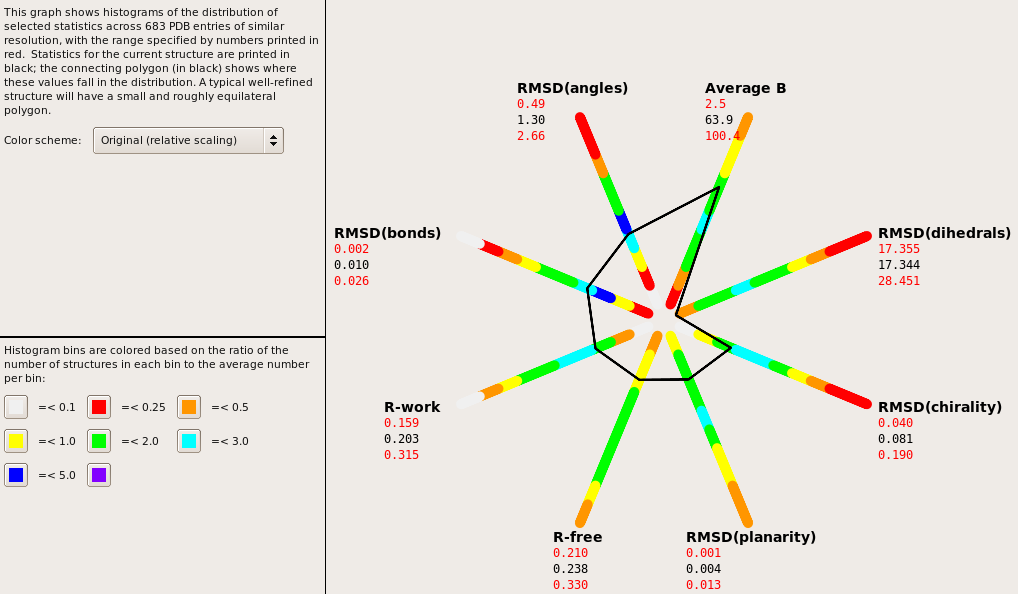
Hi all, I'm working on a ~3 angstrom structure of a protein with approximately 800 residues. The structure is mostly helical with interspersed turns. I've been using Phenix exclusively for the refinements and have a model that I believe to be pretty good with the exception of the dihedrals (see attached). I've looked at and employed some of the general suggestions for low resolution structures here: http://strucbio.biologie.uni-konstanz.de/ccp4wiki/index.php/Refinement#Refin... I tried using "discard_psi_phi=False" but it only resulted in a change dihedrals of +0.10 with minimal changes to Ramachandran outliers. I'm unsure how to proceed and would love to hear any thoughts/opinions/suggestions. Thanks for all the help! -Jon
{kind=link}

On Mar 29, 2010, at 4:46 PM, J. Fleming wrote:
I'm working on a ~3 angstrom structure of a protein with approximately 800 residues. The structure is mostly helical with interspersed turns. I've been using Phenix exclusively for the refinements and have a model that I believe to be pretty good with the exception of the dihedrals (see attached).
Actually, what that image says is that your dihedrals are "too good" - for most statistics, the closer to the center the vertices of the polygon are, the better. (A few, such as average B, don't really have "direction", although if they're outside the normal range, this is sometimes cause for concern.) I'm a little curious why the dihedrals would end up being restrained so tightly, but the statistics all look excellent, so you needn't worry. -------------------- Nathaniel Echols Lawrence Berkeley Lab 510-486-5136 [email protected]
participants (2)
-
 J. Fleming
J. Fleming -
 Nathaniel Echols
Nathaniel Echols