3Fe4S clusters in real-space refinement

Hi PHENIX BB, I'm currently refining a structure with 3Fe4S clusters into some high resolution cryoEM maps (1.57 Ang). However, refinement messes up the Fe4S cluster, by moving the existing Fe and S atoms into the wrong positions in the density. Is anyone aware of this issue and/or knows of a simple way to prevent this from happening? Cheers, Rhys -- Dr Rhys Grinter Lab Head Molecular Physiology of Microbial Pathogens (MP2) Lab Monash University +61 (0)3 9902 9213 +61 (0)403 896 767

Have you seen: Iron–sulfur clusters have no right angles Nigel W. Moriarty and Paul D. Adams, Acta Cryst. (2019). D75, 16–20 http://journals.iucr.org/d/issues/2019/01/00/rr5165/index.html The Phenix F3S library worked well for our Complex II structures a few years back. And those structures definitely confirm that the angles are not 90* for F3S. (FES Fe2S2 has close to right angles, I think) You have to be sure that the atoms are arranged in the same way as in the model or the restraints will try to invert your cluster;, and that the Cys ligands connect to the right irons. Ed Rhys Grinter wrote on 4/18/2022 12:24 AM:
Hi PHENIX BB,
I'm currently refining a structure with 3Fe4S clusters into some high resolution cryoEM maps (1.57 Ang). However, refinement messes up the Fe4S cluster, by moving the existing Fe and S atoms into the wrong positions in the density.
Is anyone aware of this issue and/or knows of a simple way to prevent this from happening?
Cheers,
Rhys
-- Dr Rhys Grinter Lab Head Molecular Physiology of Microbial Pathogens (MP2) Lab Monash University +61 (0)3 9902 9213 +61 (0)403 896 767
_______________________________________________ phenixbb mailing list [email protected] https://urldefense.com/v3/__http://phenix-online.org/mailman/listinfo/phenix... Unsubscribe: [email protected]

Rhys
You can use the phil option
superpose_ideal_ligand = *None all SF4 F3S DVT
to superpose an ideal F3S to avoid the mess. I'm also happy to take a
closer look.
Cheers
Nigel
---
Nigel W. Moriarty
Building 33R0349, Molecular Biophysics and Integrated Bioimaging
Lawrence Berkeley National Laboratory
Berkeley, CA 94720-8235
Phone : 510-486-5709 Email : [email protected]
Fax : 510-486-5909 Web : CCI.LBL.gov
ORCID : orcid.org/0000-0001-8857-9464
On Mon, Apr 18, 2022 at 11:50 AM Edward Berry
Have you seen: Iron–sulfur clusters have no right angles Nigel W. Moriarty and Paul D. Adams, Acta Cryst. (2019). D75, 16–20 http://journals.iucr.org/d/issues/2019/01/00/rr5165/index.html
The Phenix F3S library worked well for our Complex II structures a few years back. And those structures definitely confirm that the angles are not 90* for F3S. (FES Fe2S2 has close to right angles, I think) You have to be sure that the atoms are arranged in the same way as in the model or the restraints will try to invert your cluster;, and that the Cys ligands connect to the right irons. Ed
Hi PHENIX BB,
I'm currently refining a structure with 3Fe4S clusters into some high resolution cryoEM maps (1.57 Ang). However, refinement messes up the Fe4S cluster, by moving the existing Fe and S atoms into the wrong positions in
Rhys Grinter wrote on 4/18/2022 12:24 AM: the density.
Is anyone aware of this issue and/or knows of a simple way to prevent
this from happening?
Cheers,
Rhys
-- Dr Rhys Grinter Lab Head Molecular Physiology of Microbial Pathogens (MP2) Lab Monash University +61 (0)3 9902 9213 +61 (0)403 896 767
_______________________________________________ phenixbb mailing list [email protected]
https://urldefense.com/v3/__http://phenix-online.org/mailman/listinfo/phenix...
Unsubscribe: [email protected]
_______________________________________________ phenixbb mailing list [email protected] http://phenix-online.org/mailman/listinfo/phenixbb Unsubscribe: [email protected]
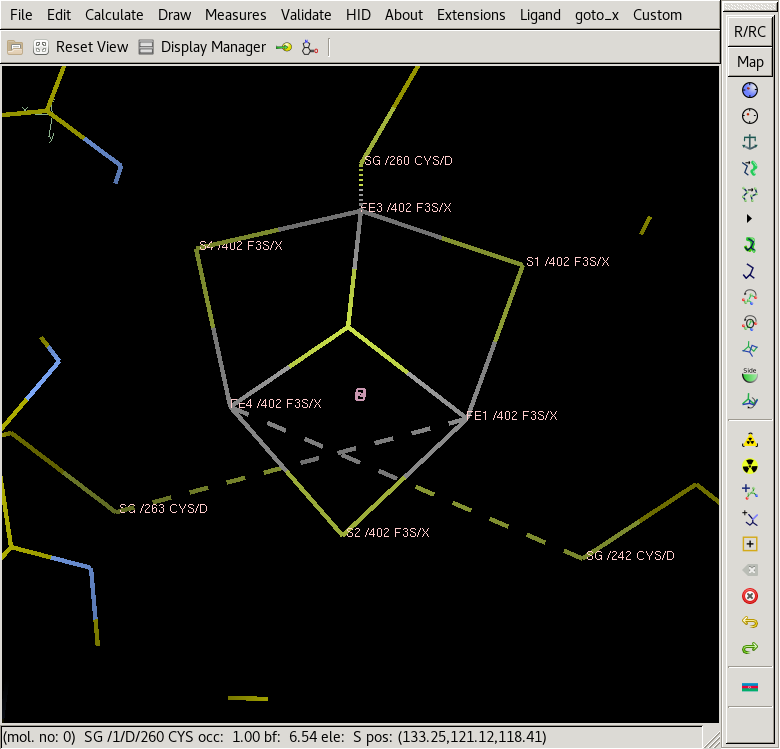
Hi Rhys, At some point the atom positions of F3S were changed, or maybe standardized, to match those of the SF4 Fe4S4 cluster. That is, if you take SF4 and remove Fe2 then you have F3S. This is good but it complicates refining some old structures, and using them to solve new structures. If you get the modern F3S (Fetch Monomer in coot) and orient it with S3 toward you, the other three S atoms S1, S2, S4 make a triangle going counterclockwise, and the Fe atoms FE1, FE3, FE4 make a smaller triangle going clockwise. If you load an older entry like 2wdq or 1yq3 both triangles go in the opposite direction. You can fix this easily in coot, although you will have to do each cluster separately. With the tool suggested by Nigel it appears they can all be fixed at once (if you are familiar with using phil options). Load the pdb you posted into coot and center on S3 of X 402 (for example) Now do regularize zone" on just residue X402 to get something like the right shape for the ligand. rotate the S3 atom toward you, and you will have something like the screenshot below. Notice the Fe atoms go counterclockwise like in the old pdbs - presumably coot optimizes bond angles starting from what you gave it, not worrying about chirality. There is a problem: the links to Cys 242 and 263 are crossed. There are two ways to fix this, and the density should tell which is right. (1) rename atoms Fe1 and Fe4 (swap the names) and don't change the link records. You should also swap S1 and S4 to keep the connectivity, although that may fix itself during refinement. In addition to uncrossing the links, this gives you the chirality of the new model. (2) rotate the F3S residue 180* abuot the vertical. This will uncross the links, but it will also put S3 behind the triangles. The density should say if this is right. This will still leave you with the old-style cluster and may get inverted in refinement, so if this second solution is correct you still need to swap Fe1/Fe4 and S1/S4; AND redefine the links for the new atom names. (NOT the pdb LINK records, but the "geometry_restraints.edits" or whatever the gui uses to define links. Or maybe auto-linking will take care of it, if the starting model is approximately right. Or, comparing the coot-regularized model with what you posted, it looks like you had Fe1 and Fe2 correct and S1 and S4 were wrong, coot moved/switched Fe1 and Fe4 to correct the connectivity but got the wrong chirality. Going back to your starting model you may only need to switch S1 and S4. Hope that helps, or, if you've already got it fixed, that it may help someone else with the same problem. Ed
{kind=link}
One correction- FES also has no right angles, according to phenix-dev-3885/modules/chem_data/geostd/f/data_FES.cif and high resolution structure 1rie.pdb Edward Berry wrote on 4/18/2022 2:45 PM:
Have you seen: Iron–sulfur clusters have no right angles Nigel W. Moriarty and Paul D. Adams, Acta Cryst. (2019). D75, 16–20 <https://urldefense.com/v3/__http://journals.iucr.org/d/issues/2019/01/00/rr5... >
The Phenix F3S library worked well for our Complex II structures a few years back. And those structures definitely confirm that the angles are not 90* for F3S. (FES Fe2S2 has close to right angles, I think) You have to be sure that the atoms are arranged in the same way as in the model or the restraints will try to invert your cluster;, and that the Cys ligands connect to the right irons. Ed
Ed
It is true that FES has no right angles. I've been meaning to write it up
so maybe now...
Cheers
Nigel
---
Nigel W. Moriarty
Building 33R0349, Molecular Biophysics and Integrated Bioimaging
Lawrence Berkeley National Laboratory
Berkeley, CA 94720-8235
Phone : 510-486-5709 Email : [email protected]
Fax : 510-486-5909 Web : CCI.LBL.gov
ORCID : orcid.org/0000-0001-8857-9464
On Sat, Apr 23, 2022 at 9:27 AM Edward Berry
One correction- FES also has no right angles, according to phenix-dev-3885/modules/chem_data/geostd/f/data_FES.cif and high resolution structure 1rie.pdb
Have you seen: Iron–sulfur clusters have no right angles Nigel W. Moriarty and Paul D. Adams, Acta Cryst. (2019). D75, 16–20 < https://urldefense.com/v3/__http://journals.iucr.org/d/issues/2019/01/00/rr5...
The Phenix F3S library worked well for our Complex II structures a few years back. And those structures definitely confirm that the angles are not 90* for F3S. (FES Fe2S2 has close to right angles, I think) You have to be sure that the atoms are arranged in the same way as in
Edward Berry wrote on 4/18/2022 2:45 PM: the model or the restraints will try to invert your cluster;, and that the Cys ligands connect to the right irons.
Ed
phenixbb mailing list [email protected] http://phenix-online.org/mailman/listinfo/phenixbb Unsubscribe: [email protected]
participants (4)
-
 Edward A. Berry
Edward A. Berry -
Edward Berry
-
 Nigel Moriarty
Nigel Moriarty -
 Rhys Grinter
Rhys Grinter