At lower resolution (lower than ~3A) refinement of biological macromolecules may be unstable. In some cases even secondary structure (SS) elements may be distorted during coordinate refinement. SS elements include helices and sheets in protein structures, base-pairs and stacking bases in nucleic acids. This applies to refinement against both X-ray and EM data. To preserve correct geometry of SS it may be useful to use specifically designed secondary structure restraints. Using SS restraints in moderate-to-low resolution refinement may help to preserve correct geometry of SS elements in the structure and sometimes correct slightly distorted SS elements.
SS restraints are available in most refinement tools in Phenix package. Their behaviour is regulated by the same scope of parameters (see the bottom of the page for the full listing). They could be turned on in GUI by checking "Use secondary structure restraints" checkbox. This is equivalent to command-line option secondary_structure.enabled=True. In this case SS elements will be found automatically and outputted to the resulting .pdb and log files. We recommend to check carefully automatic assignments of SS elements because incorrect assignments can lead to wrong refinement results.
The tutorial video is available on the Phenix YouTube channel and covers the following topics:
Secondary structure elements of proteins are helices and sheets. The main stabilizing factor is their hydrogen bonds.
The PDB format supports 10 different types of helix, but only three of these (alpha, pi, and 3_10) are common in naturally occurring proteins and have easily deciphered hydrogen-bonding rules. User-specified helices default to alpha form (hydrogen bonds between residue n and residue n+4). Sheets may be either parallel or antiparallel.
The most common way to specify secondary structure is to use HELIX and SHEET records in the beginning of .pdb file. There are several ways to obtain such records. Examples of such records:
HELIX 1 HA GLY A 86 GLY A 94 1 9 SHEET 1 A 5 THR A 107 ARG A 110 0 SHEET 2 A 5 ILE A 96 THR A 99 -1 N LYS A 98 O THR A 107 SHEET 3 A 5 ARG A 87 SER A 91 -1 N LEU A 89 O TYR A 97 SHEET 4 A 5 TRP A 71 ASP A 75 -1 N ALA A 74 O ILE A 88 SHEET 5 A 5 GLY A 52 PHE A 56 -1 N PHE A 56 O TRP A 71
Full reference to HELIX and SHEET records PDB format may be found here Hydrogen bond lengths for protein secondary structure are restrained at 2.9A with sigma 0.05A for N-O. Outliers (defined as N-O bonds greater than 3.5A) will be automatically removed, but you may prevent this with secondary_structure.protein.remove_outliers=False. This may be helpful for very poor low-resolution structures, e.g. to force proper structure on a manually built helix, but it will also cause completely spurious bond restraints to be overlooked. Three hydrogen bond angles are also restrained for alpha-helices. The first and the last bond angles are restrained with RMSD of 10 degrees, inner angles restrained with RMSD of 5 degrees. Target values are the following:
These angle restraints can be turned off by setting secondary_structure.protein.restrain_hbond_angles=False.
There are various programs available that can produce HELIX/SHEET records suitable for use in refinement. There are several ways to obtain HELIX/SHEET records for particular structure:
It is highly recommended to check manually all SS annotations coming from any source. They may often contain errors. We encourage users to verify all secondary structure annotations carefully with model. The format of HELIX/SHEET records is described here. The most common errors are: - Lack of registration part of SHEET record (second part of SHEET record when sense is not 0) - Strands are not combined into sheets, i.e. all SHEET records have different - Wrong sense in SHEET record - Not necessary pi-helices containing only 3 residues
Secondary structure elements for nucleic acids are base-pairs and stacking pairs. Base-pairs are supposed to be somewhat planar and stacking nucleobases are supposed to be parallel to each other. Several types of restraints to make these features present in resulting model are available in Phenix. None of them are symmetry-aware, e.g. for PDB ID 9dna basepair hydrogen bonds could not be determined automatically. Nevertheless, they still could be specified manually via geometry_restraints.edits mechanism.
Restraints to keep correct geometry of base-pairs involves hydrogen bonding, planarity and paralleltiy restraints. Hydrogen bond restraints include distance and angle restraints. Two types of restraints available to keep base-pairs planar: standard planarity restraints and parallelity restraint. We use parallelity by default. User can change it to planarity or even use both of them (not recommended) separately for each defined base-pair in parameter file. User also can change type of base-pairing using Saenger classes*:
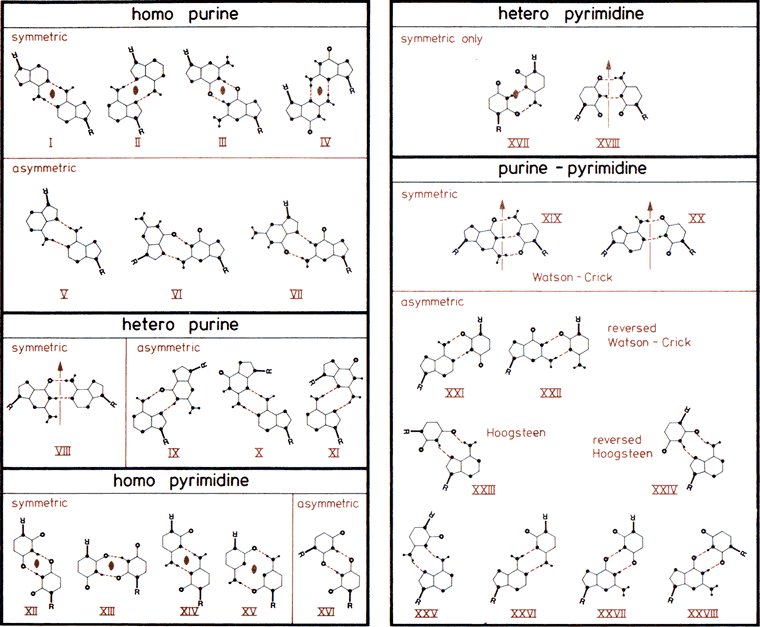
The 28 possible base-pairs for A, G, U(T), and C involving at least two (cyclic) hydrogen bonds. Hydrogen and nitrogen atoms displayed as small and large filled circles, oxygen atoms as open circles, and glycosyl bonds and thick lines with R indicating ribose C1' atom. Base-pairs are boxed according to composition and symmetry, consisting of only purine, only pyrimidine, or mixed purine/pyrimidine pairs and asymmetric or symmetric base-pairs. Symmetry elements and are twofold rotation axes vertical to and within the plane of the paper. In the Watson-Crick base-pairs XIX and XX and in base-pairs VIII and XVIII, pseudosymmetry relating only glycosyl links but not individual base atoms is observed. Drawn after compilations in (33,457).
We use parallelity restraint to keep stacked pairs parallel to each other.
The annotation could be done automatically during refinement and it will be printed out to .def file for future runs. Another way is to use phenix.secondary_structure_restraints command-line utility which can produce annotations in suitable format. PDB does not have suitable records, therefore the only way now to supply custom annotations is to use Phenix parameter files.
Documentation coming soon!