Preparing your data for TEXTALTM
TEXTALTM
takes, as input, an electron density map and automatically assigns amino
acids to the density. Only maps of 2.4 Å resolution or better have
been tested. The results have been optimized for 2.8 Å maps. Limiting the
resolution to 2.8 Å has been shown to help on the high resolution
maps which have been tested. TEXTALTM will
produce the best results if there is only one
molecule in the input map and if the amino acid sequence
is provided. However, these are not absolute requirements.
TEXTALTM is designed to fit maps rapidly
with minimal manual intervention. TEXTALTM will
only fit maps which are interpretable by humans.
MAPMAN
can be used to reformat electron density maps.
MAPMAN Authors : Gerard J. Kleywegt
& T. Alwyn Jones, Dept. of Cell and Molecular Biology, Uppsala University,
Biomedical Centre, Box 596, SE-751 24 Uppsala, SWEDEN
After installing MAPMAN, type the
following commands from the command line:
mapman
re
m1
<name of input map>
<format of input map> (type ‘?’ to see a format list)
wr
m1
<name for output map>
X-PLOR
quit
How many protein molecules are included in your electron
density map?
TEXTALTM will perform best if you submit electron
density covering only one molecule and if the molecule is centered in the
middle of the map so it is not fragmented.
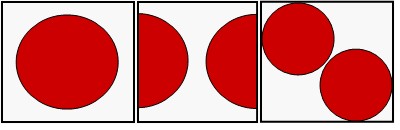
Good aaaaaaaaaaaaBad
aaaaaaaaaaaaMarginal
In some space groups it may be difficult to find one molecule and create
a map centered on that one molecule. It may help to create a mask around the
molecule and use that to pick bounds for a map, which will contain only one
molecule in one piece. The reasons for you to take the time to make a map
covering only one molecule are:
- Some of the routines in TEXTALTM run exponentially
more slowly as the map size increases.
- The sequence alignment near the end of TEXTALTM
produces unpredictable results when it receives chains for multiple molecules.
© 2002 Texas A&M University All
rights reserved