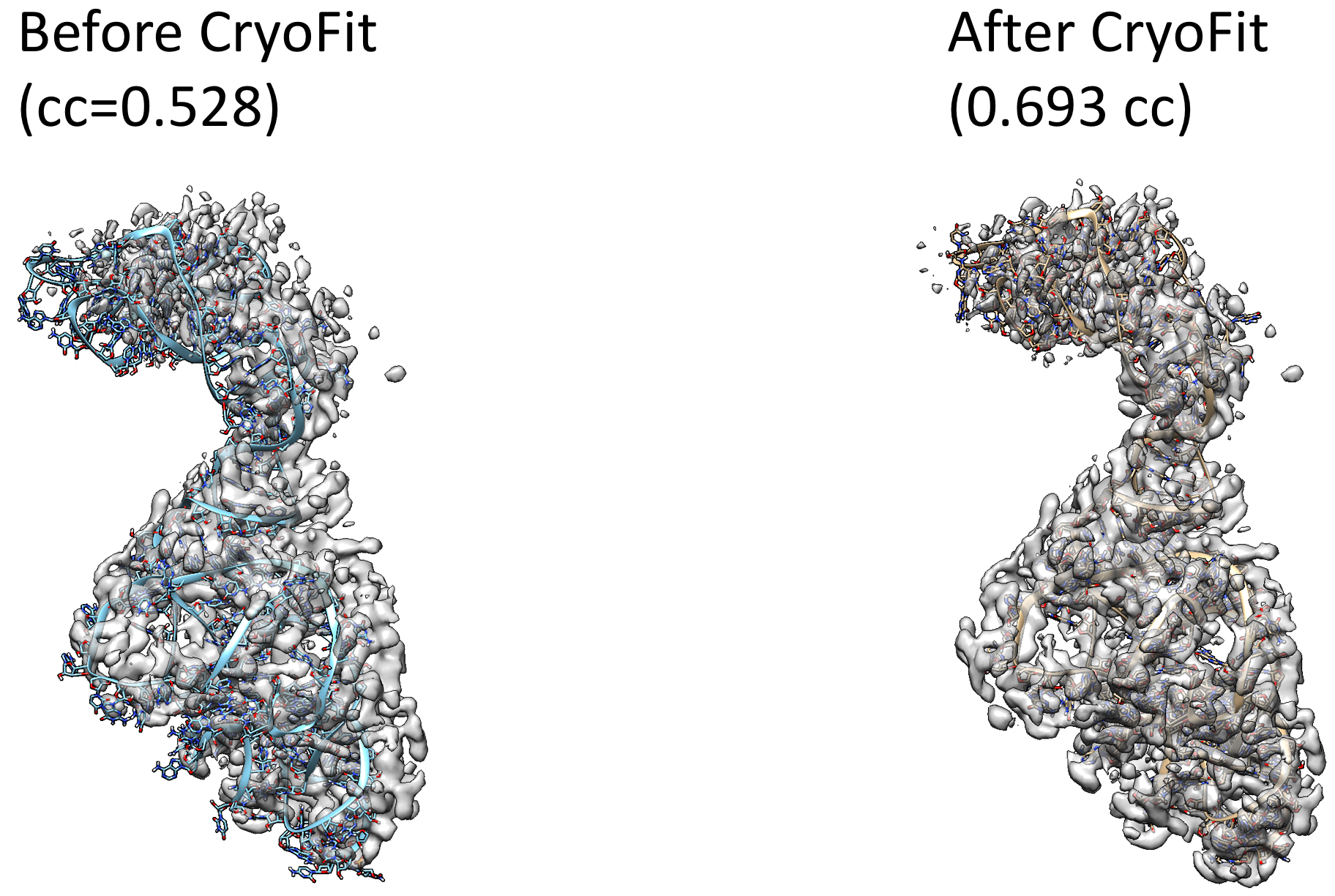
CryoFit fits biomolecule (protein and nucleic acids) to Cryo-EM map that is reconstructed by single particle analysis.
Install method, tutorial run files, troubleshooting can be found here.
<initial_model>.pdb and <target_map>.sit
cryo_fit.run can be executed via the GUI, or the command line:
% cryo_fit.run <initial_model>.pdb <target_map>.sit
example command line:
% cryo_fit.run tRNA.pdb tRNA.sit
A final cryo_fitted structure: steps/7_cryo_fit/cryo_fitted.pdb (or cryo_fitted.gro if you specified .gro for your vmd visualization)
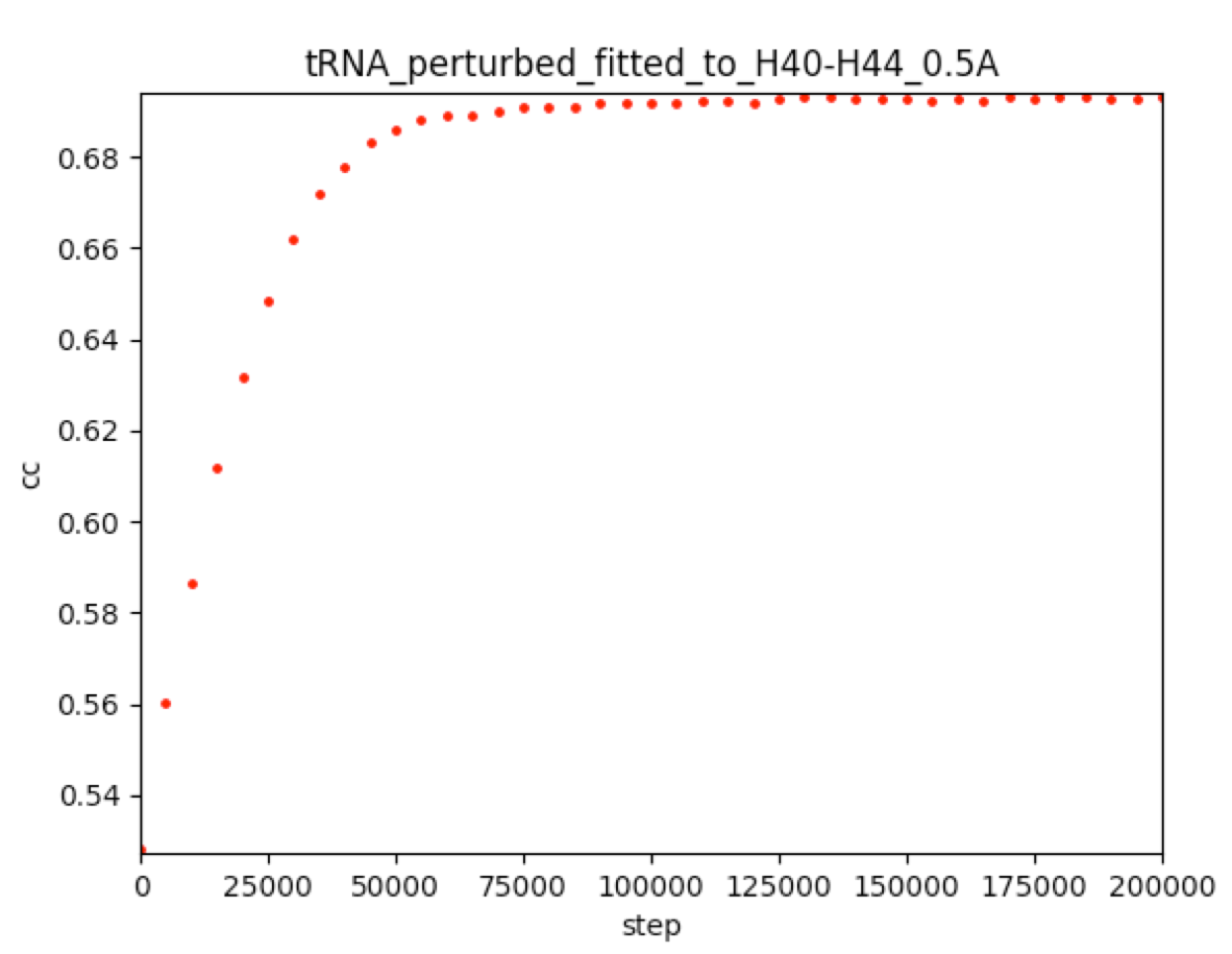
Correlation coefficients (CC) between cryo_fitted structures and cryo-EM maps: steps/7_cryo_fit/cc_record
S. Kirmizialtin, J. Loerke, E. Behrmann, C. MT. Spahn, K. Y Sanbonmatsu, Using Molecular Simulation to Model High-Resolution Cryo-EM Reconstructions, Methods Enzymol., 558, 2015, 497-514
All options will be used as default. Gromacs expert users are welcome to customize those options if they wish.
{{phil:cryo_fit.command_line.run}}