Contents
cryo_fit2 is under active development. Protein modeling is good.
However, for RNA modeling, please consider to use cryo_fit1 instead.
Pavel Afonine, Doo Nam Kim (doonam@lanl.gov)
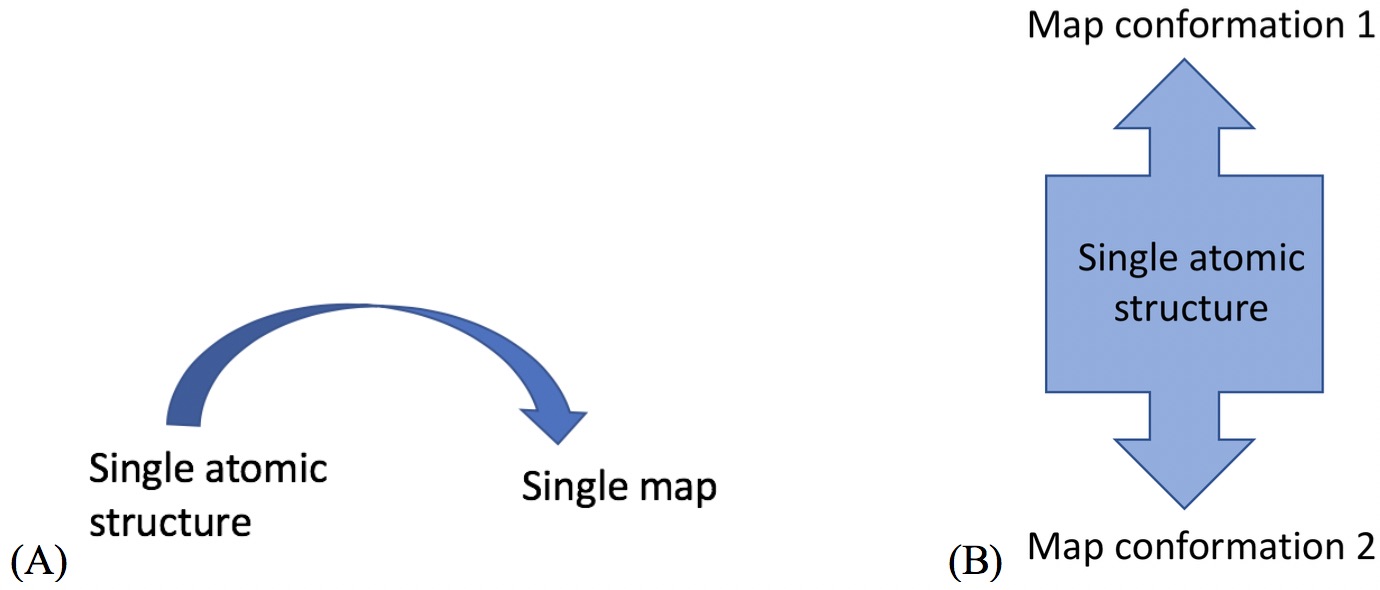
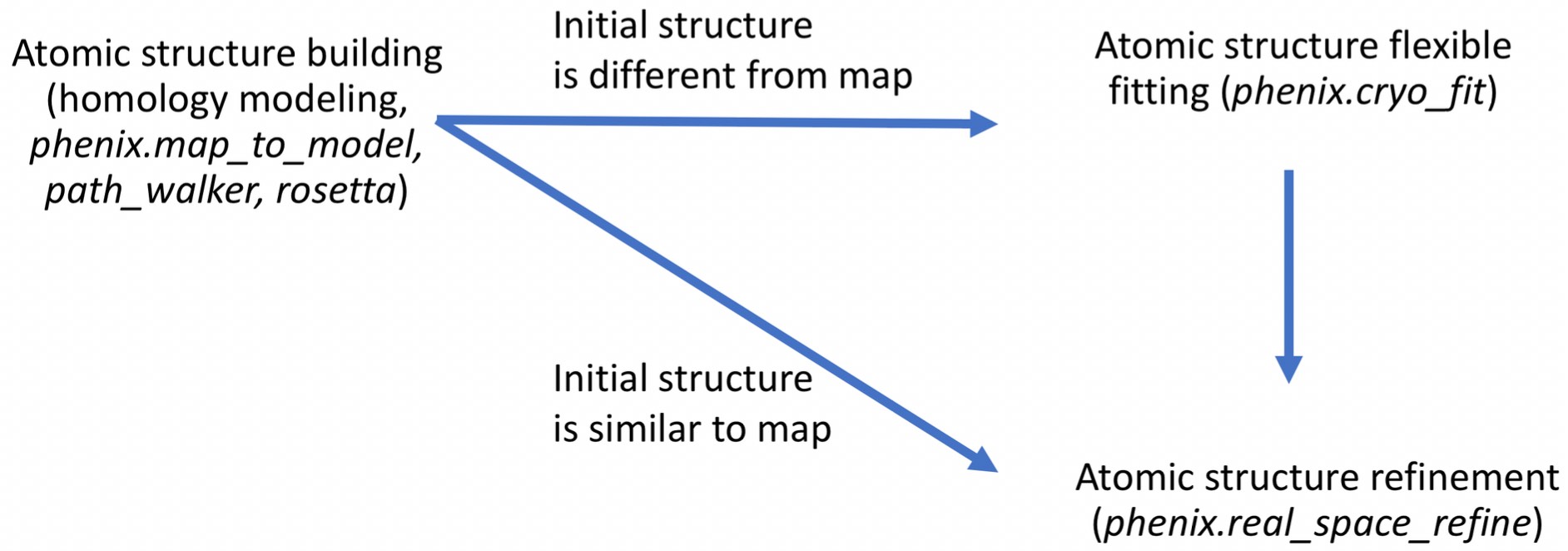
Unlike cryo_fit version 1 that uses gromacs, cryo_fit2 runs within phenix suite. Therefore, it doesn't require gromacs installation and is faster to execute. It suits the need not only traditional "static" fitting but also "dynamic" fitting. According to Doonam's benchmark, cryo_fit2 better fits than cryo_fit1 in 6 cases out of 8 cases (cryo-EM maps have 3~24 angstrom resolutions. They tie for 1 case, cryo_fit1 better fits for the last 1 case where artifically made cryo-EM map is used).
This program uses phenix dynamics written by Pavel.
Simulated annealing is carried out by default to minimize the objection function T (=T_target_map * wx + T_geom * wc ). wx is cryo-EM map weight and wc is geometry keeping weight.
See the cryo_fit2 FAQ