Contents
This tutorial will show you how to fit biomolecule atomic structures into cryo-EM maps using molecular dynamics simulation with PHENIX commandline
For GUI execution, please see the cryo_fit_gui_tutorial
Theoretical explanation of cryo_fit is here
For installation of cryo_fit, please see the installation notes for cryo_fit
<initial_model> and <target_map>
Available format: .cif and .pdb
The initial model is a guide or template structure (CIF/mmCIF/pdb) that is close to a target cryo EM map structurally.
A user can use either map_to_model or UCSF chimera (Tools -> Volume Data -> Fit in Map) to prepare the initial model.
Available format: .ccp4 and .map (MRC style in binary file) and .sit (Situs style in text file)
% phenix.cryo_fit <initial_model> <target_map>
example command line:
% phenix.cryo_fit model.pdb map.ccp4
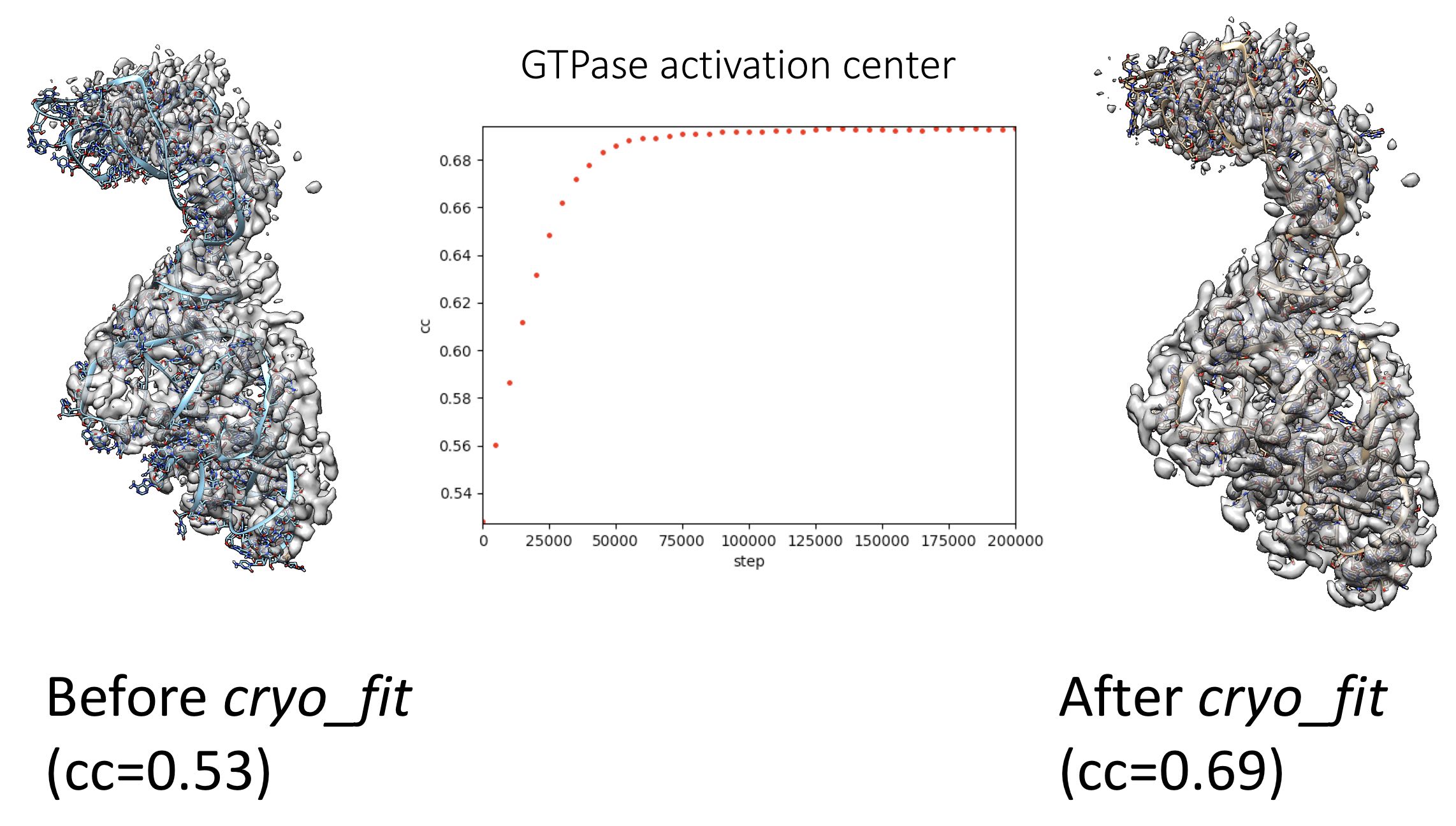
A final cryo_fitted structure: steps/8_cryo_fit/cryo_fitted.pdb (and cryo_fitted.gro for vmd visualization)
Correlation coefficients (CC) between cryo_fitted structures and cryo-EM maps: steps/8_cryo_fit/cc_record
gromacs4.5.5 seems to not handle H2O (water) heteroatom. cryo_fit will remove water molecules (if any) from the input .cif/.pdb and fit to cryo electron microscopy map.
cryo_fit doesn't handle non-canonical "residue"s such as 7C4, BMA, GDP, ILX, NAG, SEP, TRX. The cryo_fit will simply exclude those residues and report what are excluded.
S. Kirmizialtin, J. Loerke, E. Behrmann, C. MT. Spahn, K. Y Sanbonmatsu, Using Molecular Simulation to Model High-Resolution Cryo-EM Reconstructions, Methods Enzymol., 558, 2015, 497-514
All options will be used as default if unspecified. Gromacs expert users are welcome to customize those options if they wish.
| Option | Default value | Description of inputs and uses |
| emweight_multiply_by | 8 | Multiply by this number to the number of atoms for weight for cryo-EM map bias. For example, emweight = (number of atoms in gro file) x (emweight_multiply_by which is 8) The higher the weight, the stronger bias toward EM map rather than MD force field and stereochemistry preserving constraints. If user's map has a better resolution, higher value of emweight_multiply_by is recommended since map has much information. If user's map has have a worse resolution, lower value of emweight_multiply_by is recommended for more likely geometry. If CC (correlation coefficient) needs to be improved faster, higher number of emweight_multiply_by is recommended. |
| number_of_cores_to_use | max cores | Specify number of cores for minimization and cryo_fit. If it is not specified, or max is chosen, the cryo_fit will try to use most cores automatically (up to 16) |
| number_of_steps_for_cr yo_fit | None | This is the initial number of steps for cryo_fit. Eventually, cryo_fit will increase it depending on molecule size and cc trend. For tutorial files, this will be 70,000 |
| number_of_steps_for_mi nimization | None | Specify number of steps for minimization. If this is left blank, cryo_fit will estimate it depending on molecule size.number of steps for cryo_fit. Enough minimization will prevent "blow-up" during MD simulation later. |