Using the LigandFit GUI
- Overview
- Configuration
- Output
- References
Overview
LigandFit is one of the four PHENIX "wizards" for automated structure
determination. It performs flexible ligand fitting into difference maps
using RESOLVE, followed by optional real-space refinement. As elsewhere, this
documentation covers the use of the GUI specifically, and is not a
general-purpose reference for program function. See the command-line
documentation for a complete list of parameters and details
on usage.
Note that waters will be omitted when placing the ligand. However, if you
are using pre-calculated map coefficients, you may need to remove waters
beforehand to avoid "flattening" the difference density around the ligand.
Protein atoms overlapping with the ligand density will prevent a successful
placement, so some editing of the input structure is generally recommended.
Configuration
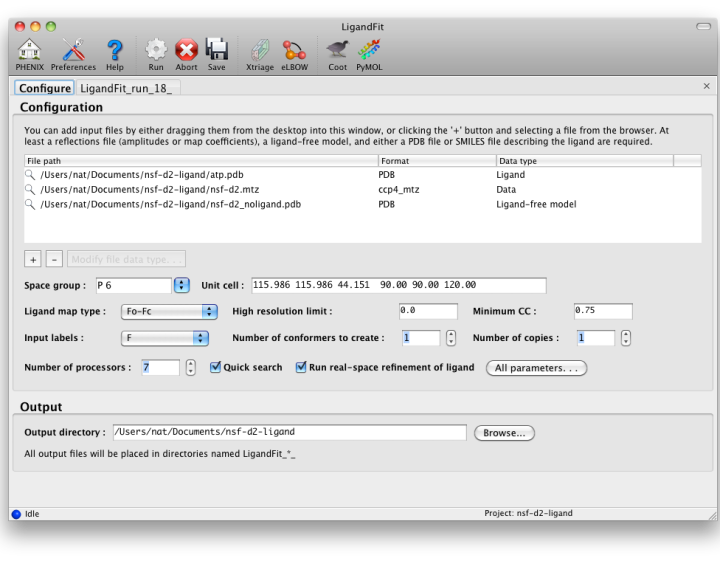
The first tab in the LigandFit window contains a field for adding input files,
which can be dragged and dropped from the desktop, or picked from a file
browser by clicking the "+" button.
To run, you will need the following input files:
- A PDB file containing the initial ligand-free model. (It is okay for this
to contain previously-placed ligands as long as the intended binding site
is empty.) Water molecules will be automatically removed if necessary.
- An MTZ file containing either experimental amplitudes (F,SIGF) or
pre-calculated difference map coefficients.
- A file describing your ligand. This should be either a PDB file containing
a single residue, or a text file containing a SMILES string. In the
latter case, eLBOW will be used to generate the starting geometry.
Additional optional files:
- A PDB file containing ligand coordinates for comparison at the end of
the program.
- A PDB file containing previously fit ligands (in addition to the starting
model), which will be used to exclude occupied regions from fitting.
- CIF files defining ligand restraints for refinement.
- A reflections file containg F/I, SIGF/SIGI, and FreeR_flag for running
a full round of phenix.refine after fitting.
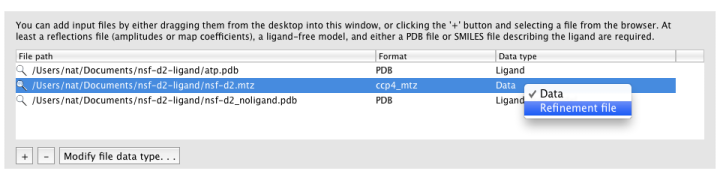
Ligandfit will try to guess what each input file should be used for
automatically. If it does not choose the desired data type, you can change
this by right-clicking the "Data type" column for that file, or selecting
the file and clicking the "Modify file data type" button below the list.
MTZ column labels (and map type, if applicable) will be extracted automatically
when a file is loaded.
By default, LigandFit runs multiple slightly different jobs in parallel and
picks the best result from these; however, check the "Quick search" box if
you want it to run a fast single job instead.
Other options:
- Ligand map type tells LigandFit what type of map to calculate or
expect in the input file. If you are supplying map coefficients from
phenix.refine or another program, "Pre-calculated" is appropriate. If
you are using raw data, "Fo-Fc" is the standard, but you may use "Fobs"
instead.
- Minimum CC sets the correlation to density required to consider a
placement successful.
- Real-space refinement will optimize the fit to density after placing
the ligand. This is optional (and may be unnecessary if you intend to
run phenix.refine immediately afterward), but is usually very fast and
improves the fit.
- Number of conformers to create determines how the ligand is partitioned
for placement of fragments. When this is set to 1, RESOLVE will try to
identify rigid groups automatically. Larger numbers will instead use
eLBOW to generate multiple conformations which are used to identify the
rigid groups. This is somewhat slower, but often more accurate.
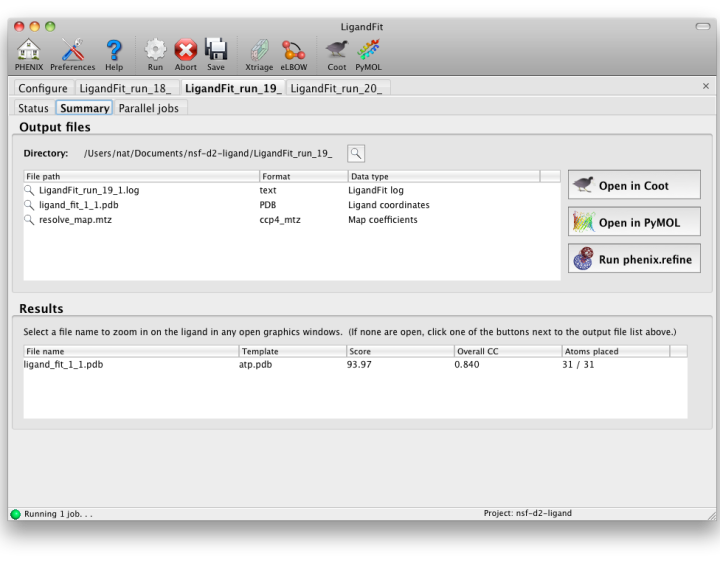
Output
LigandFit will generate a difference map used for fitting, and a separate PDB
file for each copy placed. All of these can be opened at once in one of
the graphics programs by clicking the buttons next to the file list. The
button labeled "Run phenix.refine" will launch that GUI with the start model,
reflections (if amplitudes were supplied), and all ligand output files already
loaded.
References
- Ligand identification using electron-density map correlations. T. C.
Terwilliger, P. D. Adams, N. W. Moriarty and J. D. Cohn. Acta Cryst. D63,
101-107 (2007)
- Automated ligand fitting by core-fragment fitting and extension into
density. T. C. Terwilliger, H. Klei, P. D. Adams, N. W. Moriarty and
J. D. Cohn. Acta Cryst. D62, 915-922 (2006)
|