| Python-based Hierarchical ENvironment for Integrated Xtallography |
| Documentation Home |
Density modification with multi-crystal averaging with phenix.multi_crystal_average
Author(s)
Purposephenix.multi_crystal_average is a tool for carrying out density modification, including NCS symmetry within a crystal and electron density from multiple crystals. UsageHow phenix.multi_crystal_average works:The inputs to phenix.multi_crystal_average are a set of PDB files that define the NCS within each crystal and the relationships of density between crystals, structure factor amplitudes (and optional phases, FOM and HL coefficients) for each crystal, and starting electron density maps for one or more crystals. The PDB files should each be composed of the exact same chains, placed in a different position and orientation for each NCS asymmetric unit of each crystal. You might create these PDB files by molecular replacement starting with the same search model for each crystal. You should not refine these MR solutions; they are only used to get the NCS relationships and the NCS will be more reliably found if the models for all NCS asymmetric units are identical. You can break the NCS asymmetric unit into domains and place them independently. You can specify the domains by giving them unique chain IDs, (or you can use the routine edit_chains.py to do this for you, see below). A separate NCS group will be created for each domain. Additionally if your NCS asymmetric unit consists of more than one chain (A+B for example) then each chain will always be treated as a separate NCS group. phenix.multi_crystal_average first uses the supplied PDB files to calculate NCS operators relating the NCS asymmetric unit in each crystal to all other NCS asymmetric units in that crystal and in other crystals. This is done by adding the unique chains in one crystal to each PDB file in turn, finding all the NCS relationships from all chains in that composite PDB file, and removing duplicate identity transformations. For example, suppose the NCS asymmetric unit is one chain (A,B,C....). Then to to relate all NCS asymmetric units to the NCS asymmetric unit of crystal 0, phenix.multi_crystal_average will compare all chains in the PDB file for each crystal to the unique chain in the PDB file for crystal 0, generating one NCS operator for each chain in each crystal. In this process the unique chain (in this case the NCS asymmetric unit of crystal 0) is renamed to a unique name (usually "**") and a composite PDB file is created with this chain along with all the chains in the PDB file for the crystal being considered, and phenix.simple_ncs_from_pdb is used to find the NCS operators. The centroids of the chains defining NCS are used as centers of the regions where the NCS operator is to be applied. If the supplied PDB files have more than one domain or chain in each NCS asymmetric unit, then the domains or chains are grouped into separate NCS groups. Once NCS operators have been identified, density modification is carried out sequentially on data from each crystal. During density modification for one crystal, the current electron density maps from all other crystals are used in generating target density for density modification in exactly the same way as NCS-related density is normally used when only a single crystal is available. First the asymmetric unit of NCS is defined, in this case including the density in all NCS copies within the crystal being density modified as well as the density in all NCS copies in all other crystals. The asymmetric unit of NCS is the region over which the NCS operators apply. It is assumed to be identical for all NCS copies for all crystals, with orientation and position identified by the NCS operators. It is identified as the region over which all NCS copies have correlated density. If a mask for the protein/solvent boundary is supplied (by specifying "use_model_mask"), then the asymmetric unit of NCS is constrained to be within the non-solvent region of the map. Alternatively, if you request that the domains provided in your PDB files be used to define the NCS asymmetric unit (by specifying "write_ncs_domain_pdb") then the the NCS asymmetric unit (for each NCS group) is limited to the region occupied by the corresponding chains in your PDB files. Then a target density map is created for the crystal being density modified. For each NCS copy in this crystal, the average density for all other NCS copies in this and other crystals is used as a target. Finally, statistical density modification is carried out using histograms of expected density, solvent flattening, and the NCS-based target density for this crystal. The process is then repeated for all other crystals. For those crystals for which no starting phases were available, one additional step is carried out in which the target density map is used by itself to calculate a starting electron density map (using RESOLVE map-based phasing). This entire process is carried out several times, leading to electron density maps for all crystals that typically have a high level of correlation of density within all NCS copies in each crystal and between the corresponding NCS regions in different crystals. Output files from phenix.multi_crystal_averagedenmod_cycle_1_xl_0.mtz: Density-modified map coefficients for crystal 0, cycle 1. Crystal 0 is the first crystal specified in your pdb_list, map_coeff_list, etc. denmod_cycle_5_xl_1.mtz: Density-modified map coefficients for crystal 1, cycle 5. These map coefficients are suitable for model-building. They also contain HL coefficients that can optionally be used in refinement. As the HL coefficients contain information from all crystals they may in some cases be useful in refinement (normally you would only use experimental HL phase information in refinement as the NCS-based information would come from your NCS restraints in refinement). Graphical interface
The GUI is listed in the "Maps" category of the main interface. The
configuration tab contains a set of tabs for the crystal forms:
ExamplesStandard run of phenix.multi_crystal_average:Running phenix.multi_crystal_average from the command line is easy. Usually you will want to edit a small parameter file (run_multi.eff) to contain your commands like this: # run_multi.eff commands for running phenix.multi_crystal_average
# use: "phenix.multi_crystal_average run_multi.eff"
multi {
crystal {
pdb_file= "gene-5.pdb"
map_coeffs = "resolve_1_offset.mtz"
datafile = "phaser_1_offset.mtz"
datafile_labin = "FP=F SIGFP=SIGF PHIB=PHI FOM=FOM HLA=HLA HLB=HLB HLC=HLC HLD=HLD"
solvent_content = 0.43
}
crystal {
pdb_file= "multi.pdb"
map_coeffs = "None"
datafile = "multi.pdb.mtz"
datafile_labin = "FP=FP"
solvent_content = 0.50
}
}
Then you can run this with the command: phenix.multi_crystal_average run_multi.eff In this example we have 2 crystals. Crystal 1 has starting map coefficients in crystal_1_map_coeffs.mtz and data for FP in crystal_1_data.mtz. The contents of this crystal are represented by crystal_1.pdb. The second crystal has no starting map, has data for FP as well as PHI and HL coefficients in crystal_2_data.mtz, and the contents of this crystal are represented by crystal_2.pdb. The solvent contents of the 2 crystals are 0.43 and 0.50, and 5 overall cycles are to be done. NOTE: Usually you will want to supply coefficients for a density-modified map (if available, and perhaps with NCS as well) for your starting map coefficients, and you will want to supply experimental phases and HL coefficients for your datfiles. The column label strings like "FP=FP" are optional...if you say instead "None" then phenix.multi_crystal_average will guess them for you. NOTE: You need to put quotes around the None. Also note: the order of entering pdb_list, map_coeff_list, data_file_list, solvent_content_list etc matters. They must all match. Run of phenix.multi_crystal_average with multiple domains:If your PDB files have more than one NCS domain within a chain, then you may want to split the chains up into sub-chains representing the individual NCS domains. This will provide a better definition of the NCS operators when the PDB files are analyzed. You can use the jiffy "edit_chains.py" to do this. This jiffy splits your chains up into sub-chains based on the domains that you specify in "edit_chains.dat". NOTE: edit_chains.py only works if your chains have single-letter ID's. (It simply adds another character to your chain ID's to make new ones.) If you have two-letter chain ID's, then you'll have to do this another way. To use it, type: phenix.python $PHENIX/phenix/phenix/autosol/edit_chains.py file.pdb edited_file.pdbThe file edit_chains.dat is required and should look like: A 1 321 A 322 597 A 598 750 A 751 902 A 903 1082 B 1 58 B 424 425 B 59 101 B 343 423 B 102 342where the letter and residue range is your chain ID and residue range for a particular domain. You should specify these for ALL chains in your PDB files (not just the unique ones). Run of phenix.multi_crystal_average using PDB files to define the NCS asymmetric unit:If you specify the parameter write_ncs_domain_pdb=True, then phenix.multi_crystal_average will write out domain-specific PDB files for each domain in your model (based on its analysis of NCS, one for each NCS group). Then it will use those domain-specific PDB files to define the region over which the corresponding set of NCS operators apply. This is generally a good idea if you have multiple domains in your structure. Possible ProblemsSpecific limitations and problems:
Literature
Additional informationList of all multi_crystal_average keywords
-------------------------------------------------------------------------------
Legend: black bold - scope names
black - parameter names
red - parameter values
blue - parameter help
blue bold - scope help
Parameter values:
* means selected parameter (where multiple choices are available)
False is No
True is Yes
None means not provided, not predefined, or left up to the program
"%3d" is a Python style formatting descriptor
-------------------------------------------------------------------------------
multi
crystal
pdb_file= None PDB files, one for each crystal (One pdb_file=xxx.pdb per
crystal) These should be in the same order as datafiles and
map files. They are used to identify the NCS within each
crystal and between crystals. You should create these by
placing the unique set of atoms (the NCS asymmetric unit) in
each NCS asymmetric unit of each unit cell. Normally you would
do this by carrying out molecular replacement on each crystal
with the same search model.
map_coeffs= None Mtz files with map coefficients.(One map_coeffs=xxx.mtz
per crystal). At least one crystal must have map
coefficients. Use "None" with quotes for any
crystals that do not have starting maps. NOTE: If you have
multiple NCS groups then you need map coefficients for all
crystals.
map_coeffs_labin= None Optional labin lines for mtz files with map
coefficients. (One map_coeffs_labin=my_labin per
crystal, or none at all) They look like
map_coeffs_labin=" 'FP=FP PHIB=PHIM FOM=FOMM'" Put
each set of labin values inside single quotes, and the
whole list inside double quotes. You can leave out a
labin statement for a file by putting in
"None" and the routine will guess the column
labels
datafile= None Mtz files with structure factors and optional phases and
FOM and optional HL coefficients. One datafile for each
crystal to be included (One datafile=xxx.mtz for each
crystal).
datafile_labin= None Optional labin line for mtz file (In same order as
mtz file). It can contain FP SIGFP [PHIB FOM] [HLA HLB
HLC HLD]. It looks like this: datafile_labin='FP=FP
SIGFP=SIGFP PHIB=PHIM FOM=FOMM' You can leave out a
labin statement for a file by putting in
"None" and the routine will guess the column
labels NOTE: If you supply HL coefficients they will be
used in phase recombination. If you supply PHIB or PHIB
and FOM and not HL coefficients, then HL coefficients
will be derived from your PHIB and FOM and used in phase
recombination.
solvent_content= None Solvent content (0 to 1, typically 0.5) for each
crystal (one solvent_content=xxx for each crystal).
ha_file= None Optional file, normally containing heavy atom sites or
other coordinates that are present in this structure, but not
in the others. If supplied, the density near (within 2 A) of
atoms in this file will not be transferred to other crystals.
perfect_map_coeffs= None Optional mtz files with perfect map
coefficients for comparison.
perfect_map_coeffs_labin= None Optional labin lines for mtz files with
perfect map coefficients. For comparison and
checking only. Not normally used.
averaging
cycles= 5 Number of cycles of density modification
resolution= None high-resolution limit for map calculation Default is
use all data
fill= True Fill in all missing reflections to resolution res_fill.
res_fill= None Resolution for filling in missing data (default = highest
resolution of any datafile) Default is use all data
temp_dir= "temp_dir" Optional temporary work directory
output_dir= "" Output directory where files are to be written
use_model_mask= False You can use the PDB files you input to define the
solvent boundary if you wish. These will partially
define the NCS asymmetric unit (by limiting it to the
non-solvent region) but the exact NCS asymmetric unit
will always be defined automatically (by the overlap of
NCS-related density). Note that this is different than
the command write_ncs_domain_pdb which defines
individual regions where NCS applies for each domain.
sharpen= False You can sharpen the maps or not in the
density-modification process. (They are unsharpened at the end
of the process if so). Not normally used, as an anisotropy
correction with sharpening is normally applied to all the data.
equal_ncs_weight= False You can fix the NCS weighting to equally weight
all copies.
weight_ncs= None You can set the weighting on NCS symmetry (and
cross-crystal averaging)
write_ncs_domain_pdb= False You can use the input PDB files to define
NCS boundaries. The atoms in the PDB files will be
grouped into domains during the analysis of NCS
and written out to domain-specific PDB files. (If
there is only one domain or NCS group then there
will be only one domain-specific PDB file and it
will be the same as the starting PDB file.) Then
the domain-specific PDB files will be used to
define the regions over which the corresponding
NCS operators apply. Note that this is different
than the command use_model_mask which only defines
the overall solvent boundary with your model.
mask_cycles= 1 Number of mask cycles in each cycle of density
modification
aniso
remove_aniso= True Remove anisotropy from data files before use Note:
map files are assumed to be already corrected
b_iso= None Target overall B value for anisotropy correction. Ignored if
remove_aniso = False. If None, default is minimum of (max_b_iso,
lowest B of datasets, target_b_ratio*resolution)
max_b_iso= 40. Default maximum overall B value for anisotropy
correction. Ignored if remove_aniso = False. Ignored if b_iso
is set. If used, default is minimum of (max_b_iso, lowest B
of datasets, target_b_ratio*resolution)
target_b_ratio= 10. Default ratio of target B value to resolution for
anisotropy correction. Ignored if remove_aniso = False.
Ignored if b_iso is set. If used, default is minimum of
(max_b_iso, lowest B of datasets,
target_b_ratio*resolution)
control
verbose= True verbose output
debug= False debugging output
raise_sorry= False Raise sorry if problems
coarse_grid= False You can set coarse_grid in resolve
resolve_size= 12 Size for solve/resolve
("","_giant",
"_huge","_extra_huge" or a number
where 12=giant 18=huge
resolve_command_list= None Commands for resolve. One per line in the
form: keyword value value can be optional
Examples: coarse_grid resolution 200 2.0 hklin
test.mtz NOTE: for command-line usage you need to
enclose the whole set of commands in double quotes
(") and each individual command in single
quotes (') like this:
resolve_command_list="'no_build' 'b_overall
23' "
dry_run= False Just read in and check parameter names
base_gui_dir= None GUI parameter only
simple_ncs_from_pdb
pdb_in= None Input PDB file to be used to identify ncs
temp_dir= "" temporary directory (ncs_domain_pdb will be written there)
min_length= 10 minimum number of matching residues in a segment
njump= 1 Take every njumpth residue instead of each 1
njump_recursion= 10 Take every njump_recursion residue instead of each 1 on
recursive call
min_length_recursion= 50 minimum number of matching residues in a segment
for recursive call
min_percent= 95. min percent identity of matching residues
max_rmsd= 2. max rmsd of 2 chains. If 0, then only search for domains
quick= True If quick is set and all chains match, just look for 1 NCS group
max_rmsd_user= 3. max rmsd of chains suggested by user (i.e., if called
from phenix.refine with suggested ncs groups)
maximize_size_of_groups= True You can request that the scoring be set up to
maximize the number of members in NCS groups
(maximize_size_of_groups=True) or that scoring is
set up to maximize the length of the matching
segments in the NCS group
(maximize_size_of_groups=False)
require_equal_start_match= True You can require that all matching segments
start at the same relative residue number for
all members of an NCS group, trimming the
matching region as necessary. This is required
if residue numbers in different chains are not
the same, but not otherwise
ncs_domain_pdb_stem= None NCS domains will be written to
ncs_domain_pdb_stem+"group_"+nn
write_ncs_domain_pdb= False You can write out PDB files representing NCS
domains for density modification if you want
verbose= False Verbose output
raise_sorry= False Raise sorry if problems
debug= False Debugging output
dry_run= False Just read in and check parameter names
domain_finding_parameters
find_invariant_domains= True Find the parts of a set of chains that
follow NCS
initial_rms= 0.5 Guess of RMS among chains
match_radius= 2.0 Keep atoms that are within match_radius of NCS-related
atoms
similarity_threshold= 0.75 Threshold for similarity between segments
smooth_length= 0 two segments separated by smooth_length or less get
connected
min_contig_length= 3 segments < min_contig_length rejected
min_fraction_domain= 0.2 domain must be this fraction of a chain
max_rmsd_domain= 2. max rmsd of domains
| |||||||||
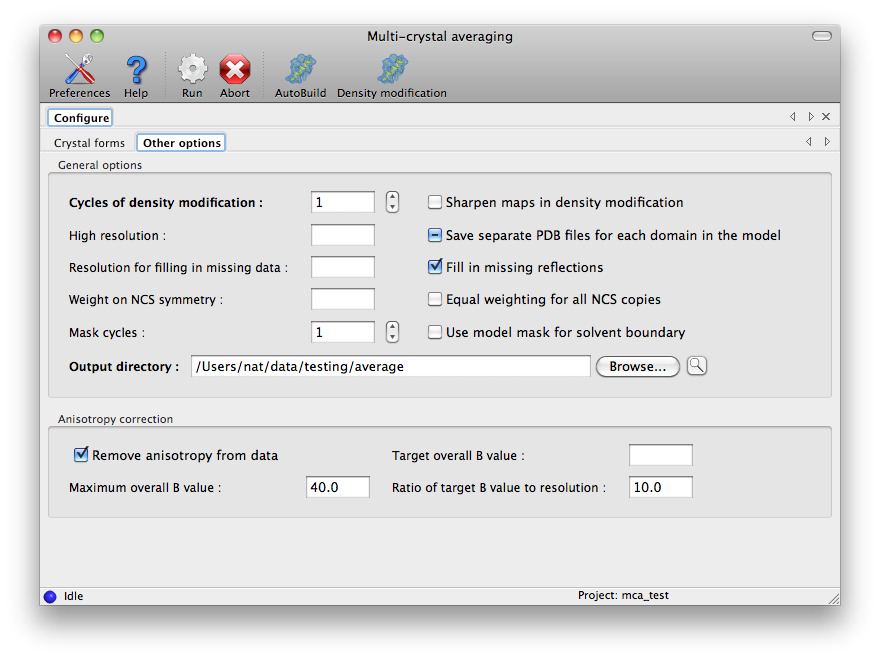
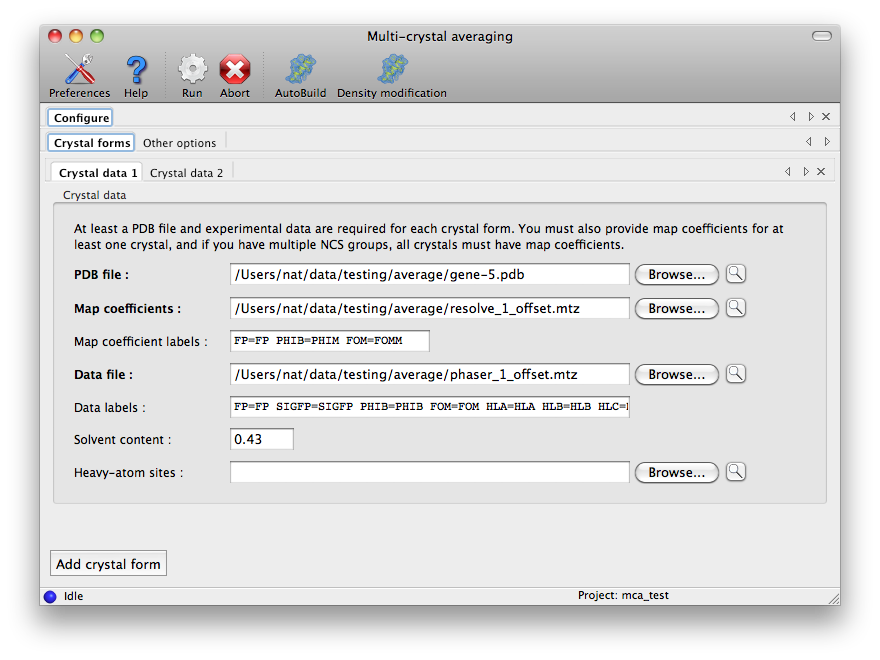 The label strings are automatically filled in when an MTZ file is loaded;
these normally do not need to be further edited. At least one crystal form
must have map coefficients, and all are required to have PDB and data files,
as well as the estimated solvent content. All other options are on the second
tab of the configuration panel:
The label strings are automatically filled in when an MTZ file is loaded;
these normally do not need to be further edited. At least one crystal form
must have map coefficients, and all are required to have PDB and data files,
as well as the estimated solvent content. All other options are on the second
tab of the configuration panel:
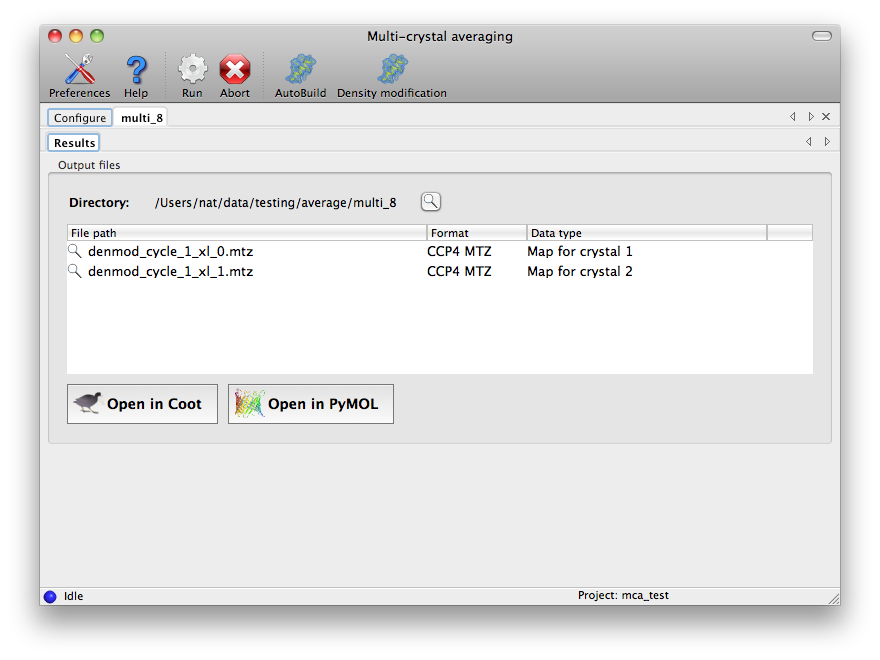
 At the end of the run, the final density-modified and averaged map coefficients
will be listed in a new tab. Selecting one of these and clicking the Coot or
PyMOL buttons below will load the map coefficients and corresponding model for
that crystal form.
At the end of the run, the final density-modified and averaged map coefficients
will be listed in a new tab. Selecting one of these and clicking the Coot or
PyMOL buttons below will load the map coefficients and corresponding model for
that crystal form.