Contents
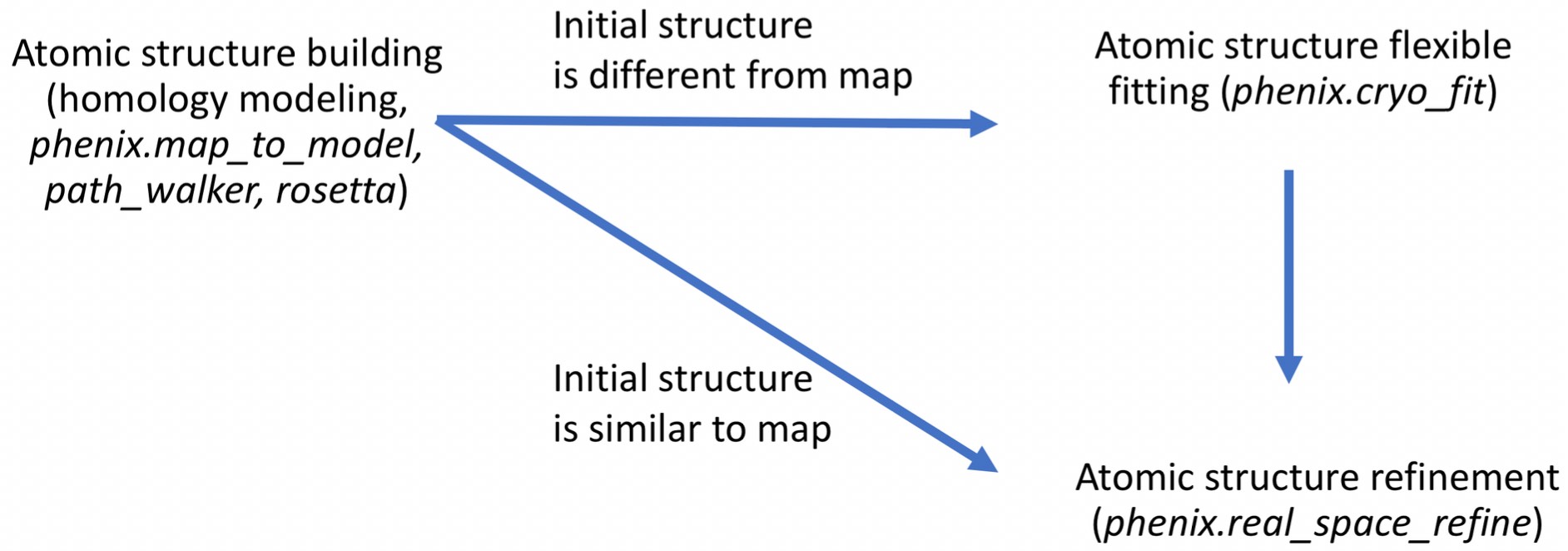
Unlike Cryo_fit1 that uses gromacs, CryoFit2 runs within phenix suite. Therefore, it doesn't require gromacs installation and is faster to execute. It suits the need not only traditional "static" fitting but also "dynamic" fitting.
This program uses phenix dynamics algorithm written by Pavel.
Simulated annealing is carried out by default to effectively minimize the objection function T (=T_target_map * wx + T_geom * wc ). wx and wc are weights.
If wx is too small like 5, it may break starting secondary structure. When wx is 100, it kept starting helix structure. If wx is too big, angle change for each step maybe too big (~30 degree), so pdb validation later (like molprobity) may raise a red flag.
We will add real_space_refine style wx, wc auto-optimization module soon.
Doo Nam Kim, Pavel Afonine