Contents
phenix.superpose_pdbs can superimpose only between pdb files that have equally/similarly aligned nucleic names. This applies to all pdb input files (not only cryo_fitted files). Consider to align both input files by "python <User Phenix Path>/modules/cryo_fit/steps/9_after_cryo_fit/align_nucleic_acid_name_into_middle/align_nucleic_acid_name_into_middle.py"
It means that the map dimensions need to be larger. (see "When cryo_fit's automatic mrc to sit map format conversion may not work properly?" in this FAQ).
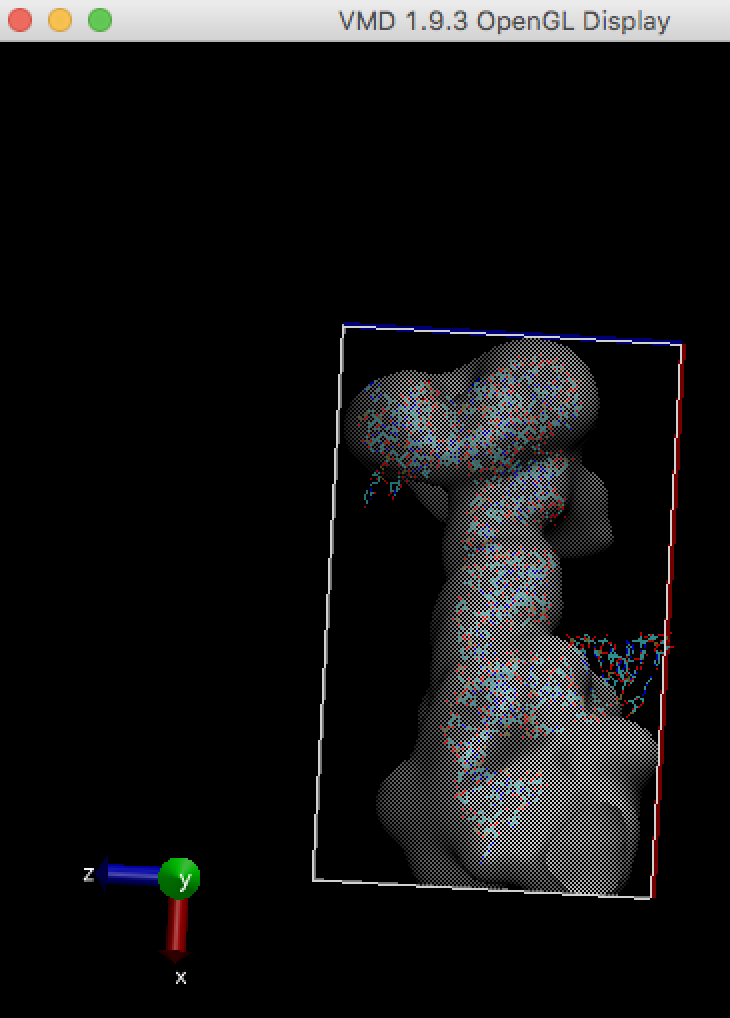
Like other MD simulations, gromacs need enough map box size to cover the atomic model to run (ziggle and wiggle). Refer Waters seems to be out of the box
For example, stuck-out red oxygen atoms outside the right edge of the box are the problem.
In order to run any MD simulation (including cryo_fit), a box should be large enough like
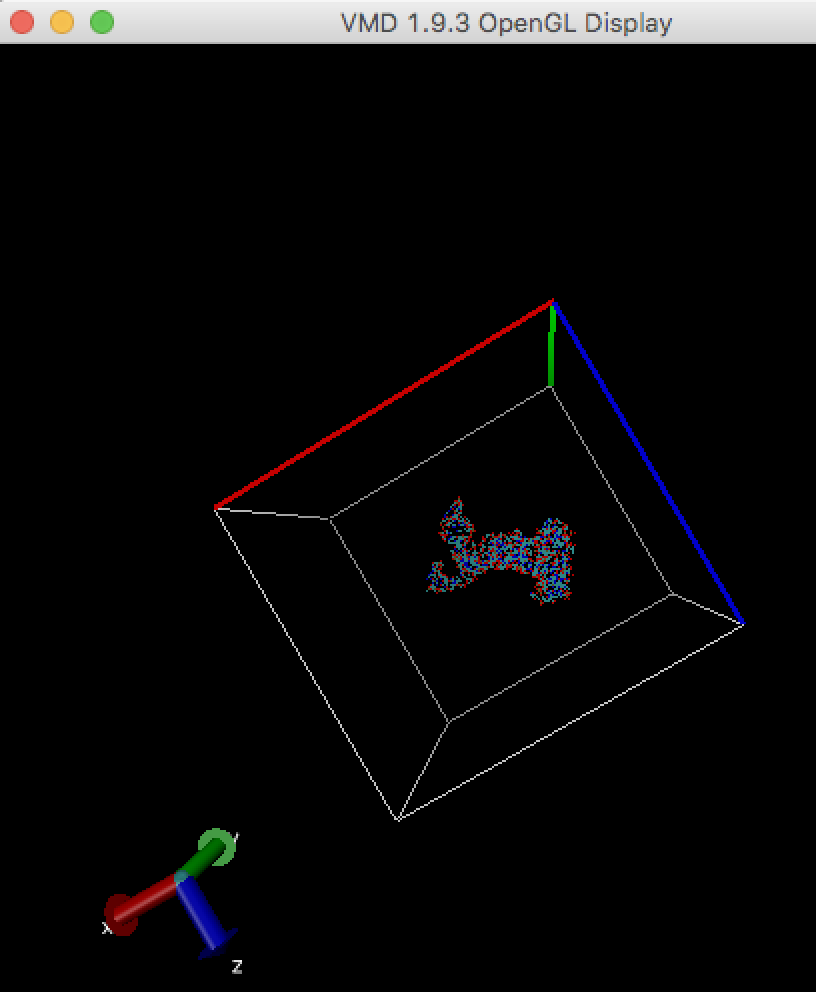
Make map box size larger (see "How to enlarge map box size?" in this FAQ), and run cryo_fit again. You can check map box size by VMD. Alternatively, remove sticking out atoms if these are unnecessary, then run cryo_fit again.
For protein modeling, I would use cryo_fit2 which is not limited by box size requirement. Most of the time, it better fits than cryo_fit1 in terms of fitting and geometry preservation anyway.