Contents
Many of the programs in Phenix, and phenix.refine in particular, allow (or require) you to specify selections of atoms in a model. Common examples include:
- TLS (constrained anisotropic displacement) and rigid-body refinement groups
- Selections for isotropic versus anisotropic B-factor refinement
- Atoms to leave out for omit map calculation
- Subset of atoms to modify properties for (in PDBTools)
- Harmonically restrain atoms during refinement
All of these parameters are defined using a common syntax similar to what is used in CNS or PyMOL. The syntax allows for selection of atomic properties such as atom name, residue number, chain ID, or B-factor, which can be combined by the use of simple boolean statements (and, or, or not).
The tutorial video is available on the Phenix YouTube channel and covers the following topics:
All atoms
all
All C-alpha atoms (not case sensitive)
name ca
All atoms with H in the name (* is a wildcard character)
name *H*
Atoms names with * (backslash disables wildcard function)
name o2\*
Atom names with spaces
name 'O 1'
Atom names with primes don't necessarily have to be quoted
name o2'
Boolean "and", "or", and "not"
resname ALA and (name ca or name c or name n or name o) chain a and not altid b resid 120 and icode c and model 2 segid a and element c and charge 2+ and anisou
Note that the and, or, and not operators have equal priority, so parentheses may be required to indicate which clauses go together. The first example above selects for all atoms with the specified names in alanine residues, but if the parentheses are omitted:
resname ALA and name ca or name c or name n or name o
it will instead select C-alpha atoms in ALA, plus all atoms named C, N, or O regardless of residue name.
Also note that the outcome of boolean expression can be somewhat different from what one may expect comparing it with natural language. For example, if one wants to select ALA and GLY residues in the molecule, the selection should be
resname ALA or resname GLY
because suitable residues have residue name ALA or GLY. On the contrary, selection
resname ALA and resname GLY
will result in empty selection because there is no residue that can satisfy both conditions simultaneously.
A single residue by number
resseq 188
Note that if there are several chains containing residue number 188, all of them will be selected. To be more specific and select residue 188 in particular chain:
chain A and resid 188
this will select residue 188 only in chain A.
A range of residues
resseq 2:10
This selects all residue numbers falling within this range, independent of their order in the PDB file. The numbering must be ascending; resseq 10:2 will not work.
Insertion codes
resid combines the residue number (resseq) with the insertion code (icode); thus the following selections are identical:
resid 188 resseq 188 and icode ' '
as are these:
resid 27C resseq 27 and icode 'C'
Specifying ordered ranges of residues
For some purposes, such as specifying TLS groups, it may be impractical to specify a numerical range of resseq attributes. This is especially the case when the residue numbering is not continuous and ascending, which is found in some PDB entries, or when the insertion code is non-blank. An alternative is to define a series of residues by their combined number and insertion code, using the through keyword:
chain A and resid 1000 through 17J
In this case, the residues will be examined in the order that they appear in the PDB file, and every atom falling between 1000 and 17J in chain A will be included. This is the only situation where the ordering of atoms is important in atom selections.
B-factors and occupancies
You may also select atoms based on their numerical properties:
bfactor > 80 bfactor = 0 occupancy < 1 occupancy = 0
Other
A few additional keywords are supported:
element H water pepnames hetero
These examples specify hydrogen atoms, water molecules (detected by residue name), common amino acid residues, and atoms labeled as HETATM, respectively.
Selecting no atoms
(not all)
The default value of most selections is None (which in the GUI will appear as a blank field), but the treatment of this differs depending on how the atom selection will be used. For omit selections, None is equivalent to (not all); for rigid-body selections, phenix.refine will default to treating the entire model as a single rigid body. For B-factor refinement, leaving the selections set to None will result in their parameterization being determined based on the input model and atom type (hydrogens are treated specially).
In the phenix.refine GUI, any valid atom selection can be visualized if you have a suitable graphics card and have already loaded a PDB file with valid symmetry information. The graphics window can be opened by clicking the "View/pick" button next to any atom selection field. Depending on the size of your structure, it may take several seconds for PHENIX to determine the atomic connectivity. The current selection, if any, will be highlighted:
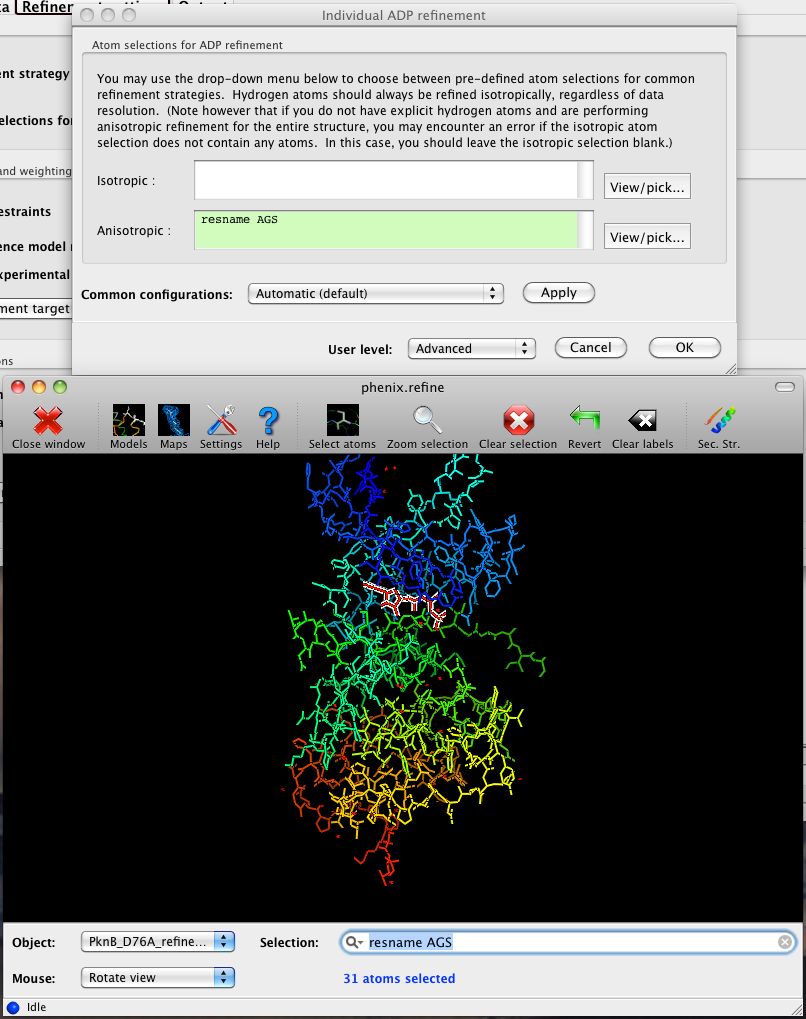
The View/pick button opens a window that allows you to type in selections and immediately visualize the results, including the number of atoms selected. On mice with at least two buttons, clicking on an atom with the right button will open a menu for selecting residue ranges or chains. However, we recommend that you learn the selection syntax, as it is much more flexible than mouse controls. Selections made in the graphics window will be sent automatically to the appropriate control.
Once this window is open, it does not need to be closed; clicking a different "View/pick" button will transfer control of the display. (On Linux, you will first need to open the Actions menu and click Restore parent window to transfer mouse and keyboard controls back to the configuration dialog.)
In some contexts (primarily in phenix.refine and PDBTools), the geometry restraints information is used to extend the allowed keywords:
resname ALA and backbone resname ALA and sidechain peptide backbone rna backbone or dna backbone water or nucleotide dna and not (phosphate or ribose) within(5, (nucleotide or peptide) backbone)
Note however that these options cannot be visually selected in the Phenix GUI at present, and they may not be recognized by all programs.