This tutorial will show you how to do comprehesive model validation within the PHENIX graphical user interface (GUI). (Validation in the PHENIX GUI reviews many of the aspects of this tutorial.)
The example used for this tutorial is called Protein Kinase A (3dnd). To get the requisite files open the GUI and set up the tutorial, as described here, called Protein Kinase A (validation). This should setup the main/GUI window. On the right-hand side click Validation and then select Comprehensive Validation (MolProbity).
In the "Comprehensive Validation" window, browse to the tutorial directory that you specified above, and select 3dnd.pdb as the "Input model", 3dnd.mtz as the "Reflections file", and 3dnd.ligands.cif as the "Restraints (CIF)" file. This CIF file defines restraints for the ligand in 3dnd. Now you are ready to run the program, select Run from the top menu bar.
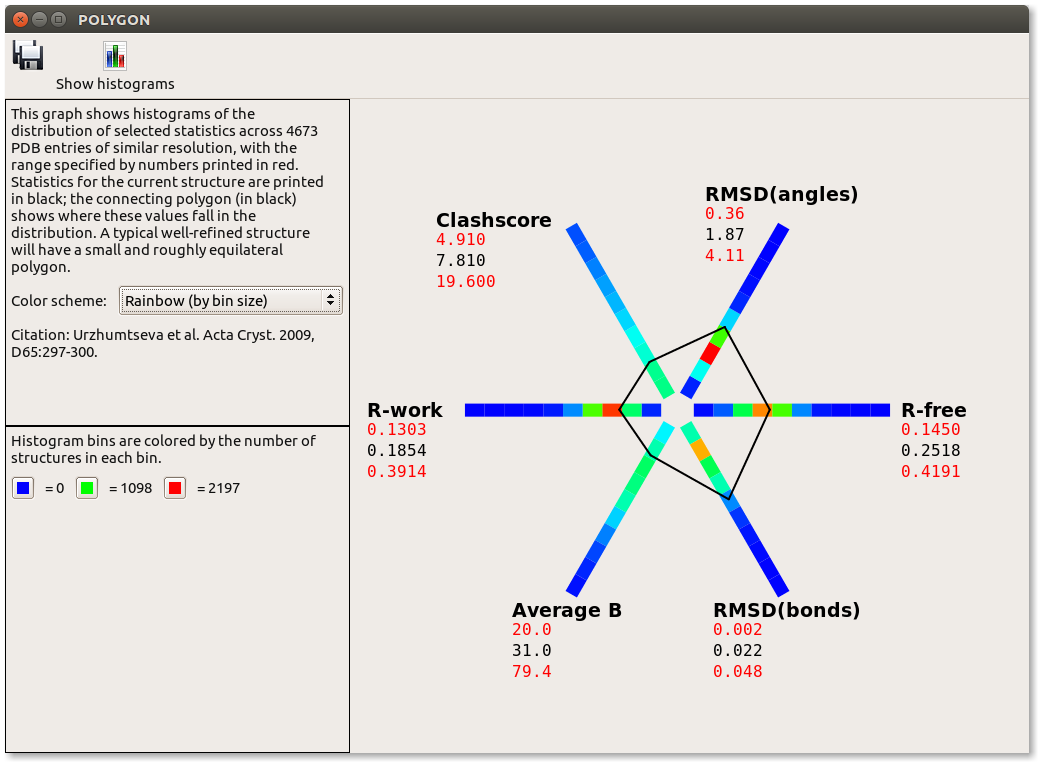
When validation has run, click on the "Compare statistics" button. This will launch a tool called "Polygon", which is used to simultaneously compare R-work, R-free, RMSD-bonds, RMSD-angles, Clashscore, and Average B factor. Well-built models will usually have a small, fairly equilateral polygon, whereas larger or significantly asymmetric deviations are indicative of model problems. As you can see for 3dnd, both RMSD-bonds and RMSD-angles are a bit large, which could indicate some misfit areas of the structure that are causing geometric strain.
Close the "POLYGON" window and return to the comprehensive validation window. Click the "Open in Coot" button. When Coot loads, note that it says "Connected to PHENIX" in the toolbar - this allows the validation GUI to communicate with Coot.
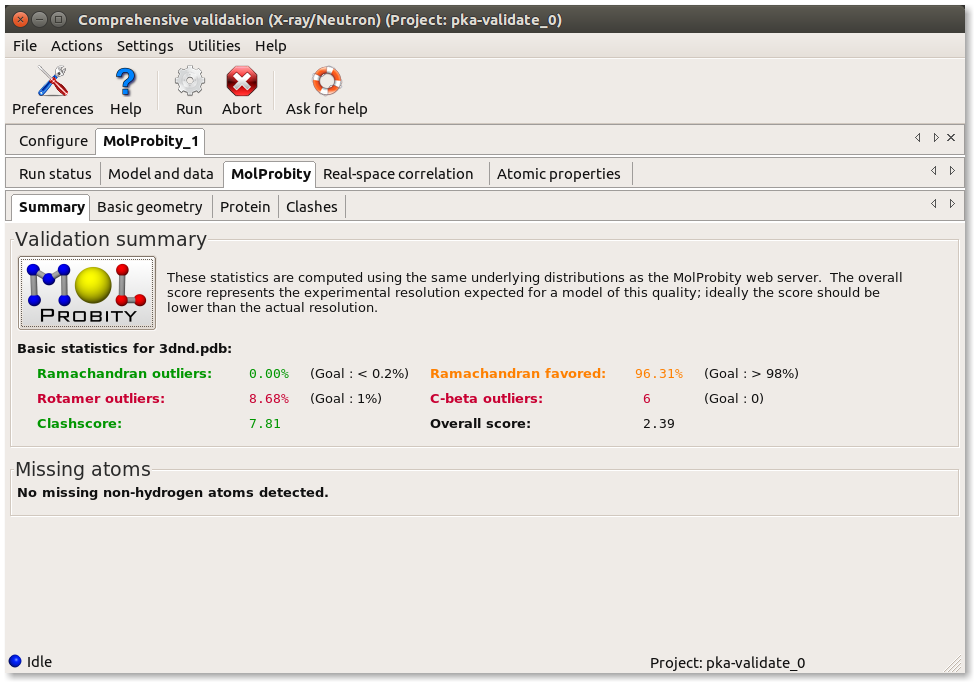
Now select the "MolProbity" tab. Under this tab, you will see sub-tabs for "Summary", "Basic geometry", "Protein", and "Clashes". If your model had RNA, there would also be an "RNA" tab. Under the "Summary" tab are most of the same overall statistics that you would find when running the MolProbity webserver. Notice the high percentage of rotamer outliers, and large number of C-beta deviations (those combine geometry problems around the Calpha into a single measure, as explained in Lovell 2003).
From the MolProbity tab, click on the "Basic geometry" tab. Here you find a summary of all bond, angle, dihedral, chirality, and planarity outliers. Outliers will be listed in the associated lists, and each item is clickable, which will center in Coot. Note the bond outlier for Ile A 163 - we'll be seeing this residue again later.
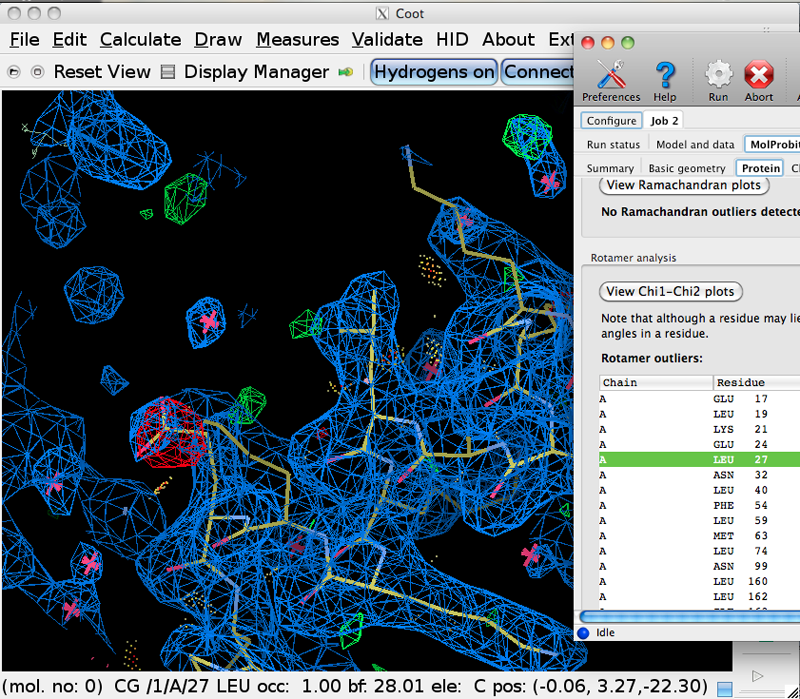
Next, go to the "Protein" tab. This section contains validation information for Ramachandran and rotamer outliers, C-beta deviations, recommended Asn/Gln/His sidechain flips (these have NOT already been done for you, as you can tell if you click on His 39 in the flip list to see it in Coot), and non-trans peptide bonds.
Find the list of rotamer outliers, and click on Leu 27 from the A chain, which will center on this residue in the Coot window. As you can see in Coot, this orientation is not a terrible fit to the density - but it is a rotamer outlier and energetically unfavorable due to an eclipsed Chi angle, and it has a suggestive difference peak. We'll use the tools in Coot to fix this sidechain. In Coot, click Calculate-> Model/Fit/Refine to bring up the window of modeling tools. First, we need to select a map, which you can do by pressing the "Select Map" button. Choose the 2FOFCWT map. Next, select "Auto Fit Rotamer", and then click on an atom in the Leu A 27 sidechain. Did you see it rotate ~180 degrees? These kinds of misfit Leu residues are common in crystal structures, but are easy to identify and fix. Change your point of view (maybe center on C-gamma) and Click "Undo" and "Redo" a few times until you are comfortable with how this change is carried out. Similarly, you can correct many of the other sidechain outliers in the GUI list.
Return to the Validation GUI window, and navigate to the list of C-beta position outliers. Select "Ile 163" from the A chain. Do you recall this sidechain from the bond-length outlier list? Misfit sidechains will often have multiple diagnostic indicators of a problem, which is useful in easily identifying the worst offenders. Notice the blob of positive density near to the sidechain, indicating that it may not be in an optimal position. Correcting misfit sidechains such as these can be tricky, as many rounds of refinement have caused distortions in the model to accommodate the misfit.
One approach that works well to fix this type of problem is to first mutate the offending residue to an alanine, and then run real-space refinement. These steps allow the C-beta position to be properly refined, without being trapped by the other misfit sidechain atoms (similar to the backrub in KiNG). To do this, select "Simple Mutate", click on an atom in Ile A 163, and then select Ala from the pop-up window. Next, select "Real Space Refine Zone", and click on 2 atoms to specify a range that runs at least 2 residues on either side of the Ile A 163 (or pick Ile 163 and hit the "a" key for autozone). Notice the subtle, but distinct movement of the C-beta position. Accept the change. Next, we'll mutate the residue back to Ile. To do this, select "Mutate and Auto Fit", click on an atom in Ile A 163, and select Ile from the pop-up menu. Notice that the newly fit sidechain is now rotated ~180 degrees, has a much better density fit for the CD1 atom, and has now placed the CG2 atom in the positive density peak. Upon further refinement, the position of this Ile will further improve, as the neighboring atoms are able to recover from the strain caused by the initial outlier.
The kind of sidechain fixups you've done in KiNG or Coot can mostly be accomplished using real-space refinement in phenix.refine (which includes a rotamer correction component), up to somewhere between 2 and 2.5 Å (and of course NQH flips are done automatically by Reduce in either MolProbity or Phenix). At lower resolution, however, real-space refinement with rotamer correction cannot reliably discern between correct and incorrect rotamers. That's a hard job for people as well, but can often be done if you have the interactive information on clashes and H-bonds from the non-pairwise, H-aware all-atom-contact dots.
Another type of outlier that can be very difficult for real-space refinement alone to resolve is an incorrect cis-peptide. The peptide bond between carbon and nitrogen joins adjacent animo acids. Due to the nearby carbonyl, the peptide bond has partial double bond character and does not rotate freely. Most peptides are trans, but genuine cis peptides occur preceding 5% of prolines and preceding about 1 in 3000 non-proline residues. Despite its rarity, a cis conformation can be tempting to model because it may appear to fit constricted or patchy density. Once modeled, cis peptides are difficult to escape, since doing do would involve rotating 180 degrees through a high energetic barrier. Fortunately, there is a tool in Coot to simplify human-directed corrections.
To get the files for this demo, open a new terminal, and navigate to the pka-validate directory. In the terminal, type "phenix.fetch_pdb --mtz 2cn3". This will fetch 2cn3 and its structure factors from the pdb, then generate an mtz file from the structure factors. Back in the Phenix window, as before, go to Validation -> Comprehensive validation (MolProbity) in the Phenix GUI. Select 2cn3.pdb as the Input model and 2cn3.mtz as the Reflections file. Then Run the validations. Once validation is complete, choose Open in Coot to get a Coot window with model and map.
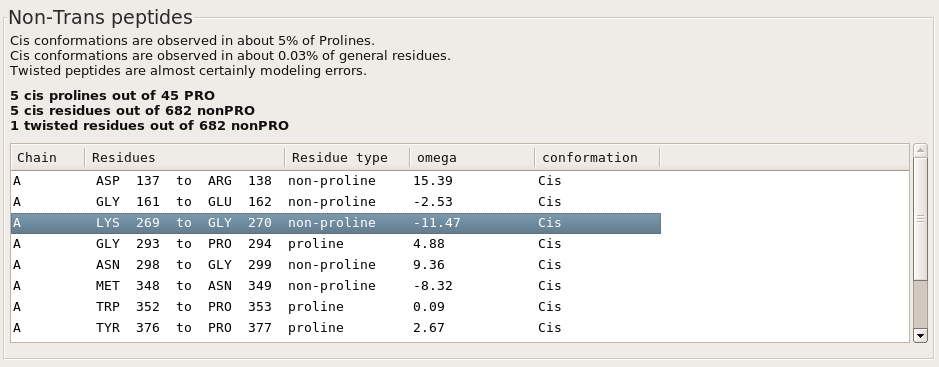
Move to the MolProbity tab of the validation output, and then the Protein tab within MolProbity. Scroll down to the bottom of the Protein tab to find Non-Trans peptides validation. Note the overall model statistics and the suspiciously high percentages of non-trans peptides in this model.
In the list of non-trans peptides, click on chain A, "LYS 269 to GLY 270" to center the Coot window on this peptide bond. [Peptide bonds necessarily span two residues, so both are listed in the validation output. However, in shorthand, peptide bonds are properly associated with their following residue, due to the special relationship cis peptide bonds have with a following proline. Thus, this location is a cis-Gly, and chain A, GLY 293 to PRO 294 would be a cis-Pro.]
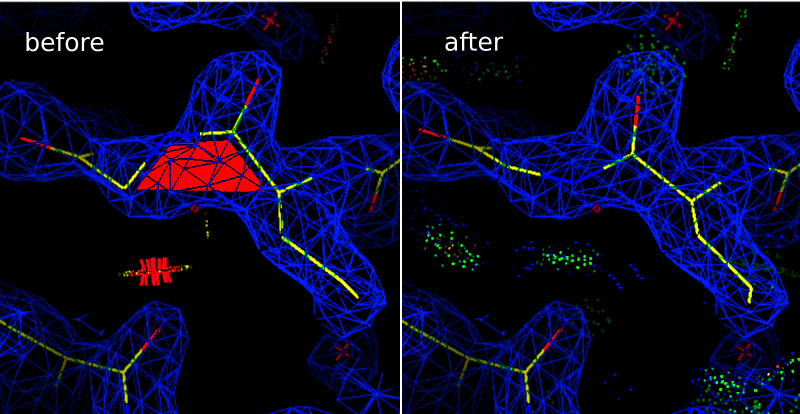
In the Coot window, observe the floating red trapezoid that marks a cis-nonProline. (The trapezoid would be green if this were a cis-Proline.) This markup makes it easy to spot cis-peptides at a glance. Also note the clash between the GLY 270 CA and the LYS 247 O.
To perform the correction: Go to Calculate -> Other Modeling Tools, and select Cis <-> Trans. Click on one of the atoms in the offending peptide bond, and Coot will flip the bond. The bond conformation has now been corrected, but the atoms have moved out of the density. Select Real Space Refine Zone, and then select atoms on either side of the peptide bond. This correction involved a large change, so be sure to give Coot at least two full residues on either side of the bond to work with. Once you have obtained a satisfactory local refinement, rerun "Local probe dots", and you should see that the LYS 247 O clash to the GLY CA has been resolved into a hydrogen bond with the GLY N.
("Local probe dots" can be added to the task bar with: Right click in task bar -> Manage Buttons -> Scroll to Validation -> check Local Probe dots)