erraser: Fang-Chieh Chou and Rhiju Das, Stanford University
ERRASER (Enumerative Real-space Refinment ASsisted by Electron-density under Rosetta) is an application for improving RNA crystal structures based on Rosetta and Phenix.
By supplementing the Rosetta RNA scoring function with electron-density restraint, ERRASER can confidently reduce the errors in RNA crystallographic models while retaining a good fit to the diffraction data. Two ERRASER applications are currently available. The standard ERRASER application remodels all the potentially problematic nucleotides in a RNA model in an automatic fashion, and output one final model. The ERRASER single residue rebuilding application apply ERRASER algorithm to a residue specified by user, and return up to 10 top-score models plus the minimized starting model. The first application is useful for automatic improving the entire model globally, and the second application is useful when the user wants to explore all possible alternative conformations for a problematic nucleotide and examine each one manually.
To run ERRASER, you must have installed Rosetta, software developed from the Baker laboratory at the University of Washington.
See the central installation notes for Rosetta
input_model.pdb: A PDB file with your starting RNA model. Please ensure it is in the standard PDB format otherwise ERRASER might not run properly.
mapfile.ccp4: A 2mFo-DFc density map for your model in CCP4 format that covers the entire unit cell. Rfree reflections should be removed in map creation to avoid overfitting. We also suggest one to fill the missing data with calculated data to avoid Fourier truncation error. The following options are suggested if PHENIX map calculation utility (GUI) is used in map creation: "Fill missing f obs", and "Exclude free r reflections" set to True. Also in "Map region", select "Unit Cell".
Running ERRASER is easy, and can be done both through the Phenix GUI or the command line. In the GUI, the program is listed under the Refinement category. Most configuration options are shown in the main window:
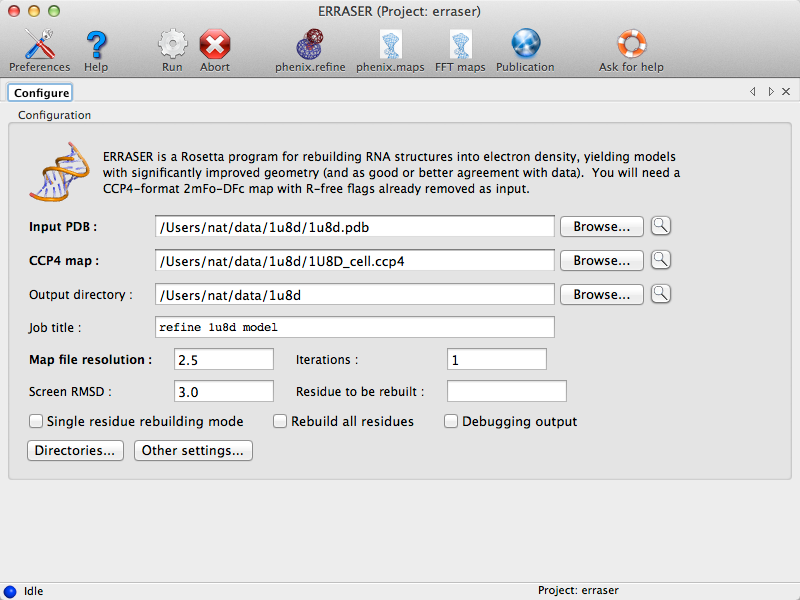
Toolbar buttons to launch phenix.maps and the FFT utility are also displayed.
From the command-line you can type:
phenix.erraser model.pdb mapfile.ccp4
This command will have erraser rebuild your entire model.
Here are some commonly used options to include: "map_reso=2.2" gives tha map resolution (2.2 angstrom here), which highly recommended to include as this usually gives better performance. "n_iterate=2" allows ERRASER to iterate twice before output the model. This takes much longer time but might gives better improvement. "fixed_res= A20 B15" gives residues to be fixed during remodeling. The format is chain ID followed by residue numbers. ERRASER currently did not model ligands (anything starts with "HETATM" in PDB, including modifies bases), proteins, and crystal contacts in a structure. Therefore we suggest to fix the position of nucleotides that are in close contact with protein or ligand component.
You can also run ERRASER in single residue rebuilding mode:
phenix.erraser model.pdb mapfile.ccp4 single_res_mode=True rebuild_res_pdb=A30
This will rebuild just residue 30 in chain A and output up to 10 models for manual inspection. (In the GUI, this is equivalent to checking the box labeled "Single residue rebuilding mode" and entering the residue ID in the field labeled "Residue to be rebuilt".)
For standard ERRASER:
model_erraser.pdb: A PDB file with your rebuilt RNA model.
For single residue rebuilding mode: model_0.pdb, model_1.pdb...: Up to 10 different models with the specified residue rebuilt.
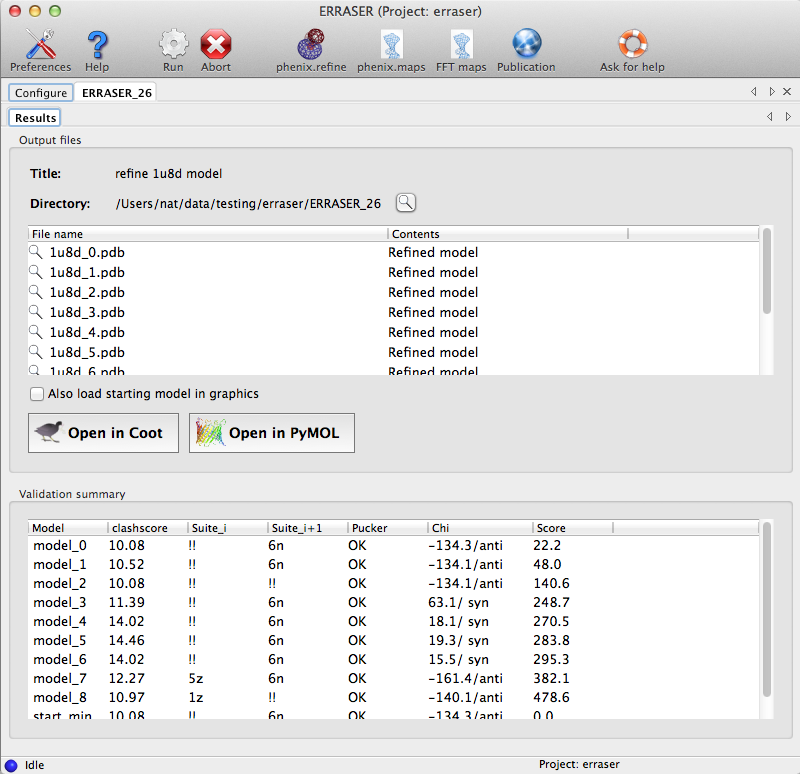
In both modes, after the models are generated, the application will analyze the models and output a detailed comparison of the changes introduced by ERRASER. The GUI displays a list of output files and a summary of validation statistics:
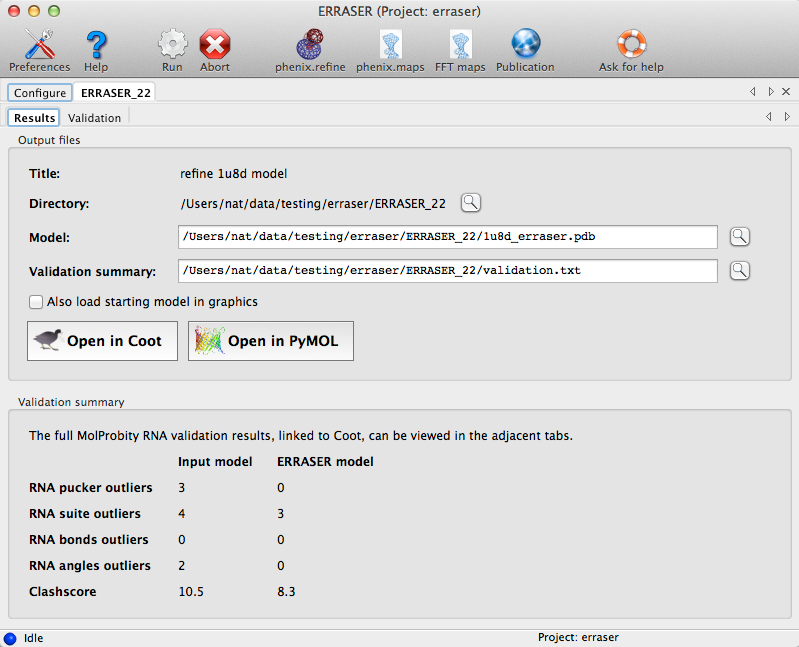
For the standard rebuilding mode, a subset of the MolProbity analyses will be displayed in additional tabs. Lists of outliers are interactive with Coot (if installed); see the validation documentation for more information. In single-residue mode, a summary of the validation criteria for the specific residue being rebuilt is shown instead:
ERRASER works only for RNA currently. Other parts in crystallographic model, including proteins, modified bases and ligands, are not being modeled. Remodeling of RNA residues that are in close contact with these components may be problematic.
Currently crystal contacts are not being modeled, which is known to cause problems in a few test cases when RNA is interacting strongly with its crystal-packing partner (ex. base-pairing and base-stacking). Right now this problem can be resolved by mannually adding the crystal-packing partner into the starting pdb file or forcing these residues as "fixed_res" during the run.