whether my following data are acceptable

Dear All, Following is the data range of my x-ray statistics by resolution bin: FOM between 0.84 to 0.86 Phase error between 17.90 to 24.14 scale factor from 0.88 to 1.07 SigmaA from 0.85 to 0.95 #work from 2688 to 2922 #test from 133 to 157 Do these data are within the acceptable range? Cheers, Dialing

Hi Dialing, you may want to have a look at this: http://www.phenix-online.org/presentations/latest/pavel_validation.pdf to learn about assessment of model and data quality, and model-to-data fit.
FOM between 0.84 to 0.86 Phase error between 17.90 to 24.14
This doesn't tell me a lot, apart from a light feeling that they look rather good than bad.
scale factor from 0.88 to 1.07
This is an arbitrary number, can be any, like 0.0123 or 123 or 1000, etc.
#work from 2688 to 2922 #test from 133 to 157
What is this? Pavel
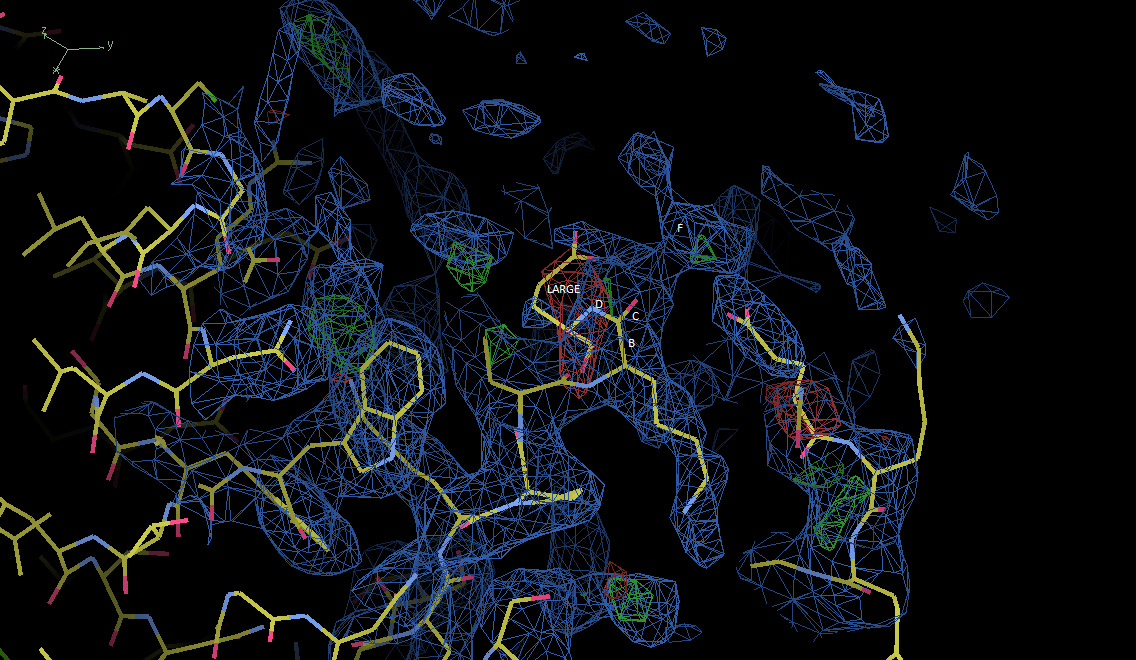
Dear All, You see the part labelled "D and "LARGE" in the attached image is the part for the last residue in a chain, however it localizes in the red part. If I can change the part labelled "D" and "LARGE" into the part labelled "F", the red should have disapeared. But after several rounds of try by Coot, I have not make the above exchange. Will you please give me your suggestions on how to change the part labelled with "D and "LARGE" into the part labelled by "F" with Coot or with some other methods? Cheers, Dialing
{kind=link}

Does the part at D connect to the polypeptide to right through region F? If so you can move each residue so its backbone is in density and spaced appropriately to connect structurally, and use regularize to connect. You have to rebuild the part on right, too. On the right, put the mainchain, not sidechain of that acidic residue into density. Are there any structural homologues of what you are building? Search pdb sequences for sequence homology and superimpose. They can give good hints for rebuilding I am amazed you have gotten so far with this with bulletin board help. Is there anyone nearby you could go to? Having personal guidance would be very helpful with coot. Tim On Dec 25, 2011, at 9:25 PM, Dialing Pretty wrote:
Dear All,
You see the part labelled "D and "LARGE" in the attached image is the part for the last residue in a chain, however it localizes in the red part. If I can change the part labelled "D" and "LARGE" into the part labelled "F", the red should have disapeared.
But after several rounds of try by Coot, I have not make the above exchange.
Will you please give me your suggestions on how to change the part labelled with "D and "LARGE" into the part labelled by "F" with Coot or with some other methods?
Cheers,
Dialing
_______________________________________________ phenixbb mailing list [email protected] http://phenix-online.org/mailman/listinfo/phenixbb
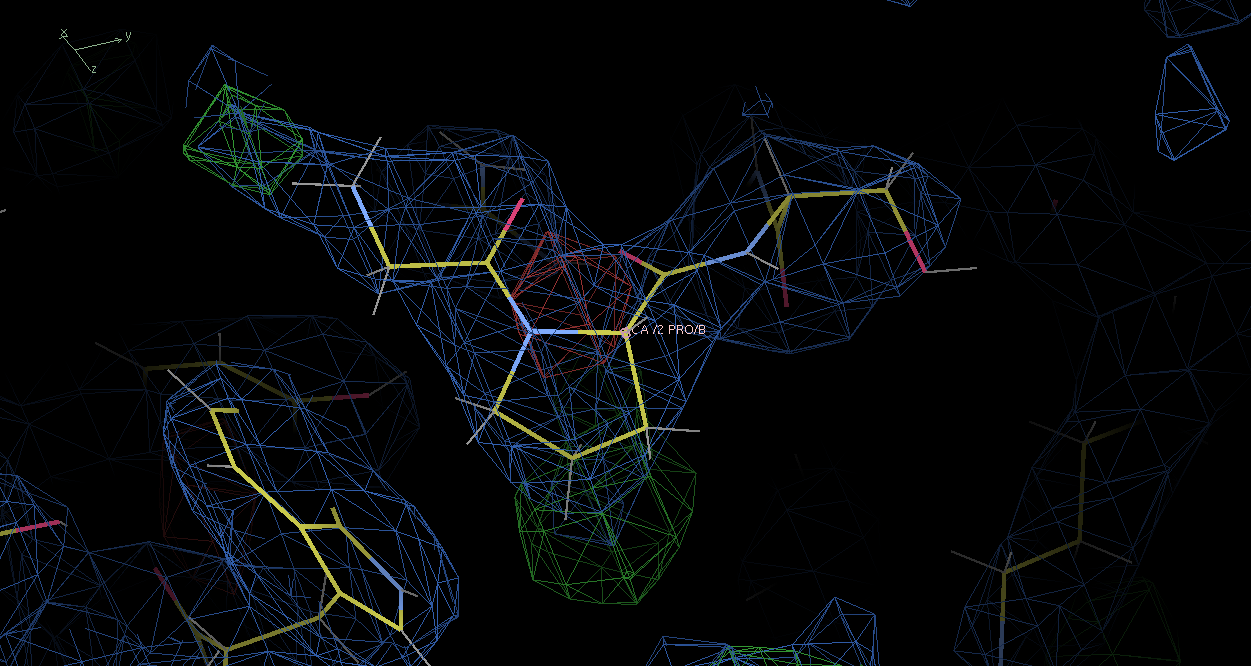
Dear All, Attached is a part of electronic density pap I assigned to Pro. After Phenix refinement, we can see under the Pro ring there is a rather large green, above the Pro ring there is a part of red. Do you think whether I assign the Pro correctly? I am looking forward to getting your reply. Cheers, Dialing
{kind=link}
{kind=link}
Hi Dialing, ideas to explore: - what's the blue density? Is it 2mFo-DFc "missing-Fo-filled" map or not? compare both. - sequence register error? - what's the level of residual map you show and what's the solvent content (may be you need to contour at higher levels?)? If solvent content is high you may need to contour residual map at higher than traditional 3sigma. Pavel On 1/1/12 5:11 PM, Dialing Pretty wrote:
Dear All, Attached is a part of electronic density pap I assigned to Pro. After Phenix refinement, we can see under the Pro ring there is a rather large green, above the Pro ring there is a part of red. Do you think whether I assign the Pro correctly? I am looking forward to getting your reply. Cheers, Dialing
_______________________________________________ phenixbb mailing list [email protected] http://phenix-online.org/mailman/listinfo/phenixbb

On Sun, Jan 1, 2012 at 5:11 PM, Dialing Pretty
Attached is a part of electronic density pap I assigned to Pro. After Phenix refinement, we can see under the Pro ring there is a rather large green, above the Pro ring there is a part of red.
Do you think whether I assign the Pro correctly?
My guess is no, but it's difficult to be certain from a single image. If you're certain that the sequence assignment of the rest of the chain is correct (not out of register), the Pro might be a mistake (in which case you would need to have the expression construct or gene re-sequenced). I should repeat what Tim Springer told you a week or two ago: it would be wise to find a more experienced crystallographer in your department/institution (hopefully there is one available!), and have him or her go through the structure with you interactively in Coot. The bulletin boards are great resources if you need a second opinion, or require help figuring out how to use the software - however, they are not a substitute for direct assistance when you're learning out how to build a model manually. We're always happy to try to answer questions like this, of course, but I think you'll find this a much slower and less effective way to learn than talking to someone personally. -Nat
Hi Nat,
Maybe you are correct. At the original Pro position, there is almost no electronic density (out of the major electronin density body). According to the opinion of Paul from Coot, the good method would be, after I build the main peptide and do the refinemnt, I add the extra rsidues (for example the Pro we just discussed) at the terminal of the major peptide just refined according to the electronic density map exposed after refinemnt.
But here I have another question and I do not know whether you can answer. For the above method introduced by Paul, after the initial refinement of the major peptide, more electronic density map could be exposed by the refinement, and we can base the newly exposed electronic density map to rebuild the extra residues.
However for rotamer, after refinement, we change the conformation of a rotamer a little, so that the rotamer would be not a outlier. Of course in this situation, after the rotamer changes into the non-outlier, I find the non-outlier rotamer does not not fit the original electronic density map so nicely. Then I do another phenix refinemnt, I hope the electronic density may would fit to the new non-outlier rotamer. However I find the electronic density may does not change at all by further refinemnt, so it means the conformation of the non-outlier rotamer is not correct.
Will you please explain to me why in the situation of Paul, new electronin density map can occur, but for my non-outlier rotamer situation, the electronic density does not change anymore by refinemnt to suit the non-outlier rotamer?
Cheers,
Dialing
From: Nathaniel Echols
Attached is a part of electronic density pap I assigned to Pro. After Phenix refinement, we can see under the Pro ring there is a rather large green, above the Pro ring there is a part of red.
Do you think whether I assign the Pro correctly?
My guess is no, but it's difficult to be certain from a single image. If you're certain that the sequence assignment of the rest of the chain is correct (not out of register), the Pro might be a mistake (in which case you would need to have the expression construct or gene re-sequenced). I should repeat what Tim Springer told you a week or two ago: it would be wise to find a more experienced crystallographer in your department/institution (hopefully there is one available!), and have him or her go through the structure with you interactively in Coot. The bulletin boards are great resources if you need a second opinion, or require help figuring out how to use the software - however, they are not a substitute for direct assistance when you're learning out how to build a model manually. We're always happy to try to answer questions like this, of course, but I think you'll find this a much slower and less effective way to learn than talking to someone personally. -Nat _______________________________________________ phenixbb mailing list [email protected] http://phenix-online.org/mailman/listinfo/phenixbb
Dear All, It says "wc is 1.0 by default and used to turn the restraints off". Then what it will mean for wc=0.0? And how about wc=150? Cheers, Dialing
On Mon, Jan 2, 2012 at 1:52 AM, Dialing Pretty
It says "wc is 1.0 by default and used to turn the restraints off". Then what it will mean for wc=0.0? And how about wc=150?
The wording of the documentation is a little confusing - 1.0 means leave the geometry restraints alone, 0 means turn them off, 150 means weight them very strongly. -Nat
P.S.: Let Phenix Autobuild rebuild the model and see what it places there. I guess Autobuild has a model building mode such that it builds whatever fits best even if that is not what's in the sequence file. Check manual to make sure you use that mode. Pavel On 1/1/12 5:11 PM, Dialing Pretty wrote:
Dear All, Attached is a part of electronic density pap I assigned to Pro. After Phenix refinement, we can see under the Pro ring there is a rather large green, above the Pro ring there is a part of red. Do you think whether I assign the Pro correctly? I am looking forward to getting your reply. Cheers, Dialing
_______________________________________________ phenixbb mailing list [email protected] http://phenix-online.org/mailman/listinfo/phenixbb
participants (4)
-
 Dialing Pretty
Dialing Pretty -
 Nathaniel Echols
Nathaniel Echols -
 Pavel Afonine
Pavel Afonine -
 Timothy Springer
Timothy Springer