
I am in the process of solving a series of protein/ligand complex structures and ran the Polygon tool on the series. I noticed that, in general, even when all the other statistics were good, the B(sol) term was a bit too high according to Polygon. (The structures with poorer statistics also had high B(sol) values as well.) Since this is a series of multiple structures of the same protein, should this be a concern for me? And if so, what are typical causes of a high B(sol) value? Matt Merski Shoichet Group UCSF

On Dec 15, 2009, at 11:53 PM, Matthew Merski wrote:
I am in the process of solving a series of protein/ligand complex structures and ran the Polygon tool on the series. I noticed that, in general, even when all the other statistics were good, the B(sol) term was a bit too high according to Polygon. (The structures with poorer statistics also had high B(sol) values as well.) Since this is a series of multiple structures of the same protein, should this be a concern for me? And if so, what are typical causes of a high B(sol) value?
I erred in making B(sol) one of the default parameters displayed in Polygon - it is obtained from bulk solvent correction and is not terribly informative by itself, and it will not change very much with refinement. There is some natural variation in this value and it's a property of the crystal rather than the model (mostly; Pavel would know for sure). A higher-than-average B(sol) is nothing to worry about as long as it's not an extreme value and your other statistics are okay. If all of your structures form similar or identical crystal lattices (very common, if no major conformational changes are involved), I would expect the B(sol) to be similar for each dataset. (The same applies to K(sol) - unless it's close to zero, it isn't very interesting.) -------------------- Nathaniel Echols Lawrence Berkeley Lab 510-486-5136 [email protected]

Dear Matt,
I noticed that, in general, even when all the other statistics were good, the B(sol) term was a bit too high according to Polygon. (The structures with poorer statistics also had high B(sol) values as well.)
Couldyou give a little bit more information so that people do not needmaketheir guess and consider multiple options ? :-) Inparticular : At which resolution do you work ? What is the percent of solvent ? With which kind of macromolecules do you work (proteing, nucleic acids, complexes, something else...)? Which refinement program do you use (I suppose that phenix.refine) ? Which are the Ksol, Bsol values (that make you warry) ? Maybesome more information that can be useful at your point of view...(forexample, a pdf-copy of your polygon would be useful; better to seeoncethan...). I think you can write to me and to Pavel directly (notto overload thephenixbb by work discussions and work files) and if wefix the problemyou will report the answer to everybody. Best regards, Sacha
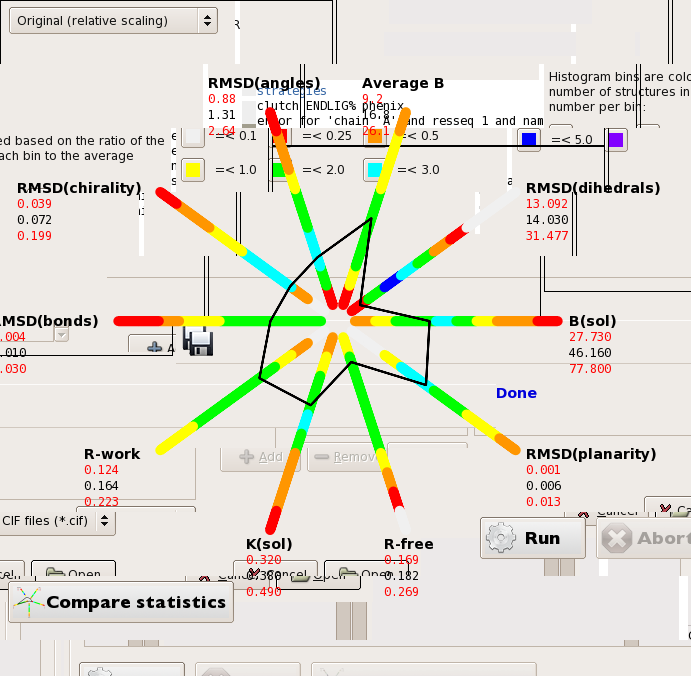
Ok. The resolution is between 1.39 and 1.9 Ang for the different crystals all in P21 21. I have been using phenix.refine to do the refinement and they are all a series of lysozyme structures with ligands. I am currently having a problem with getting the polygons to save as images. Right now they have a background which is comprised of the other parts of the image such as the name of the file and other gobletygook (see attached). Matt
{kind=link}
On Dec 16, 2009, at 6:57 PM, Matthew Merski wrote:
Ok. The resolution is between 1.39 and 1.9 Ang for the different crystals all in P21 21. I have been using phenix.refine to do the refinement and they are all a series of lysozyme structures with ligands. I am currently having a problem with getting the polygons to save as images. Right now they have a background which is comprised of the other parts of the image such as the name of the file and other gobletygook (see attached).
Is this using the save button on the toolbar, or a screen capture? I'll take a look tomorrow - this looks like a problem with your OS, however. -------------------- Nathaniel Echols Lawrence Berkeley Lab 510-486-5136 [email protected]
It was using the save button on the screen. And yes I do think its an OS
problem, but our IT has been putting out some hardware fires recently and I
saw something on the boards that there was a problem in the latest release,
so I haven't been able to track it down yet.
Matt
On Thu, Dec 17, 2009 at 2:29 AM, Nathaniel Echols
Ok. The resolution is between 1.39 and 1.9 Ang for the different crystals all in P21 21. I have been using phenix.refine to do the refinement and they are all a series of lysozyme structures with ligands. I am currently having a problem with getting the polygons to save as images. Right now they have a background which is comprised of the other parts of
On Dec 16, 2009, at 6:57 PM, Matthew Merski wrote: the image such as the name of the file and other gobletygook (see attached).
Is this using the save button on the toolbar, or a screen capture? I'll take a look tomorrow - this looks like a problem with your OS, however.
-------------------- Nathaniel Echols Lawrence Berkeley Lab 510-486-5136 [email protected]
_______________________________________________ phenixbb mailing list [email protected] http://phenix-online.org/mailman/listinfo/phenixbb
Dear All,after looking together with Matt and Pavel at the numbers sent by Matt, we agreed that everything was fine. The whole polygon is fine, all the reported values are in the "green" or "light blue" zones meaning that they are "frequent values".The value of Bbulk , equal to 46 A2, is also "very frequent". In fact, this value corresponds to the center of the (Ksolv, Bsolv) distribution reported by Andrei Fokine in ActaCryst D., 2002, vol. 58 (9), pp. 1387-1392. Therefore, even slightly higher values are not bad either; nothing to warry about.Best regards,Matt, Pavel and Sacha
participants (3)
-
 Alexandre OURJOUMTSEV
Alexandre OURJOUMTSEV -
 Matthew Merski
Matthew Merski -
 Nathaniel Echols
Nathaniel Echols