| Python-based Hierarchical ENvironment for Integrated Xtallography |
| Documentation Home |
Structure refinement in PHENIX
phenix.refine is the general purpose crystallographic structure refinement program Graphical interfaceA complete graphical interface for phenix.refine is available; it includes integration with several refinement-related utilities such as phenix.ready_set, phenix.simple_ncs_from_pdb, and phenix.find_tls_groups. Essentially all of the program details described in this document should apply to the GUI as well. Available features
Current limitations
Remark on using amplitudes (Fobs) vs intensities (Iobs) Although phenix.refine can read in both data types, intensities or amplitudes, internally it uses amplitudes in nearly all calculations. Both ways of doing refinement, with Iobs or Fobs, have their own slight advantages and disadvantages. To our knowledge there is no strong points to argue using one data type w.r.t. another. phenix.refine organizationA refinement run in phenix.refine always consists of three main steps: reading in and processing of the data (model in PDB format, reflections in most known formats, parameters and optionally cif files with stereochemistry definitions), performing requested refinement protocols (bulk solvent and scaling, refinement of coordinates and B-factors, water picking, etc...) and finally writing out refined model, complete refinement statistics and electron density maps in various formats. The figure below illustrates these steps: 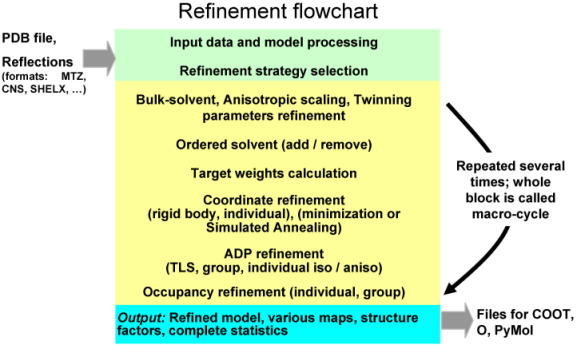 The second central step encompassing from bulk solvent correction and scaling to refinement of particular model parameters is called macro-cycles and repeated several times (3 by default). Multiple refinement scenario can be realized at this step and applied to any selected part of a model as illustrated at figure below: 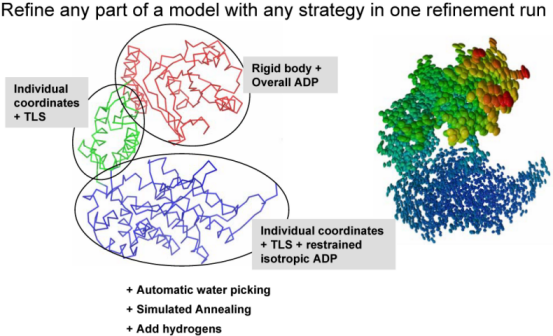 Running phenix.refinephenix.refine can be run from the command line: % phenix.refine <pdb-file(s)> <reflection-file(s)> <monomer-library-file(s)> When you do this a number of things happen:
To get information about command line options type: % phenix.refine --help To have the program generate the default input parameters without running the refinement job (e.g. if you want to modify the parameters prior to running the job): % phenix.refine --dry_run <pdb-file> <reflection-file(s)> If you know the parameter that you want to change you can override it from the command line: % phenix.refine data.hkl model.pdb xray_data.low_resolution=8.0 \ simulated_annealing.start_temperature=5000 Note that you don't have to specify the full parameter name. What you specify on the command line is matched against all known parameters names and the best substring match is used if it is unique. To rerun a job that was previously run: % phenix.refine --overwrite lysozyme_refine_001.def The --overwrite option allows the program to overwrite existing files. By default the program will not overwrite existing files - just in case this would remove the results of a refinement job that took a long time to finish. To see all default parameters: % phenix.refine --show-defaults=all Giving parameters on the command line or in filesIn phenix.refine parameters to control refinement can be given by the user on the command line: % phenix.refine data.hkl model.pdb simulated_annealing=true However, sometimes the number of parameters is large enough to make it difficult to type them all on the command line, for example: % phenix.refine data.hkl model.pdb refine.adp.tls="chain A" \ refine.adp.tls="chain B" main.number_of_macro_cycles=4 \ xray_data.high_resolution=2.5 wxc_scale=3 wxu_scale=5 \ output.prefix=my_best_model strategy=tls+individual_sites+individual_adp \ simulated_annealing.start_temperature=5000 The same result can be achieved by using: % phenix.refine data.hkl model.pdb custom_par_1.params where the custom_par_1.params file contains the following lines: refinement.refine.strategy=tls+individual_sites+individual_adp refinement.refine.adp.tls="chain A" refinement.refine.adp.tls="chain B" refinement.main.number_of_macro_cycles=4 refinement.input.xray_data.high_resolution=2.5 refinement.target_weights.wxc_scale=3 refinement.target_weights.wxu_scale=5 refinement.output.prefix=my_best_model refinement.simulated_annealing.start_temperature=5000 which can also be formatted by grouping the parameters under the relevant scopes (custom_par_2.params):
refinement.main {
number_of_macro_cycles=4
}
refinement.input.xray_data.high_resolution=2.5
refinement.refine {
strategy = *individual_sites \
rigid_body \
*individual_adp \
group_adp \
*tls \
occupancies \
group_anomalous \
none
adp {
tls = "chain A"
tls = "chain B"
}
}
refinement.target_weights {
wxc_scale=3
wxu_scale=5
}
refinement.output.prefix=my_best_model
refinement.simulated_annealing.start_temperature=5000
and the refinement run will be: % phenix.refine data.hkl model.pdb custom_par_2.params The easiest way to create a file like the custom_par_2.params file is to generate a template file containing all parameters by using the command phenix.refine --show-defaults=all and then take the parameters that you want to use (and remove the rest). % phenix.refine data.hkl model.pdb comments_in_params_file.params where comments_in_params_file.params file contains the lines:
refinement {
refine {
#strategy = individual_sites rigid_body individual_adp group_adp tls \
# occupancies group_anomalous *none
}
#main {
# number_of_macro_cycles = 1
#}
}
refinement.target_weights.wxc_scale = 1.5
#refinement.input.xray_data.low_resolution=5.0
In this example the only parameter that is used to overwrite the defaults is target_weights.wxc_scale and the rest is commented. Refinement scenariosThe refinement of atomic parameters is controlled by the strategy keyword. Those include: - individual_sites (refinement of individual atomic coordinates) - individual_adp (refinement of individual atomic B-factors) - group_adp (group B-factors refinement) - group_anomalous (refinement of f' and f" values) - tls (TLS refinement = refinement of ADP through TLS parameters) - rigid_body (rigid body refinement) - occupancies (occupancy refinement: individual, group, group constrained) - none (bulk solvent and anisotropic scaling only) Below are examples to illustrate the use of the strategy keyword as well as a few others. Refinement with all default parameters% phenix.refine data.hkl model.pdb This will perform coordinate refinement and restrained ADP refinement. Three macrocycles will be executed, each consisting of bulk solvent correction, anisotropic scaling of the data, coordinate refinement (25 iterations of the LBFGS minimizer) and ADP refinement (25 iterations of the LBFGS minimizer). At the end the updated coordinates, maps, map coefficients, and statistics are written to files. Refinement of coordinatesphenix.refine offers three ways of coordinate refinement:
All types of coordinate refinement listed above can be used separately or combined all together in any combination and can be applied to any selected part of a model. For example, if a model contains three chains A, B and C, than it would require only one single refinement run to perform SA refinement and minimization for atoms in chain A, rigid body refinement with two rigid groups A and B, and refine nothing for chain C. Below we will illustrate this with several examples. The default refinement includes a standard set of stereo-chemical restraints ( covalent bonds, angles, dihedrals, planarities, chiralities, non-bonded). The NCS restrains can be added as well. Completely unrestrained refinement is possible. The total refinement target is defined as: Etotal = wxc_scale * wxc * Exray + wc * Egeom where: Exray is crystallographic refinement target (least-squares, maximum-likelihood, or any other), Egeom is the sum of restraints (including NCS if requested), wc is 1.0 by default and used to turn the restraints off, wxc ~ ratio of gradient's norms for geometry and X-ray targets as defined in (Adams et al, 1997, PNAS, Vol. 94, p. 5018), wxc_scale is an 'ad hoc' scale found empirically to be ok for most of the cases.
Refinement of atomic displacement parameters (commonly named as ADP or B-factors)An ADP in phenix.refine is defined as a sum of three contributions: Utotal = Ulocal + Utls + Ucryst where Utotal is the total ADP, Ulocal reflects the local atomic vibration (also named as residual B) and Ucryst reflects global lattice vibrations. Ucryst is determined and refined at anisotropic scaling stage. phenix.refine offers multiple choices for ADP refinement:
All types of ADP refinement listed above can be used separately or combined all together in any combination (except TLS+individual anisotropic) and can be applied to any selected part of a model. For example, if a model contains six chains A, B, C, D, E and F than it would require only one single refinement run to perform refinement of: - individual isotropic ADP for atoms in chain A, - individual anisotropic ADP for atoms in chain B, - grouped B with one B per all atoms in chain C, - TLS refinement for chain D, - TLS and individual isotropic refinement for chain E, - TLS and grouped B refinement for chain F. Below we will illustrate this with several examples. Restraints are used for default ADP refinement of isotropic and anisotropic atoms. Completely unrestrained refinement is possible. The total refinement target is defined as: Etotal = wxu_scale * wxu * Exray + wu * Eadp where: Exray is crystallographic refinement target (least-squares, maximum-likelihood, ...), Eadp is the ADP restraints term, wu is 1.0 by default and used to turn the restraints off, wxu and wxu_scale are defined similarly to coordinates refinement (see Refinement of Coordinates paragraph). It is important to keep in mind:
Occupancy refinementList of facts about occupancy refinement in phenix.refine:
f' and f'' refinementIf the structure contains anomalous scatterers (e.g. Se in a SAD or MAD experiment), and if anomalous data are available, it is possible to refine the dispersive (f') and anomalous (f") scattering contributions (see e.g. Ethan Merritt's tutorial for more information). In phenix.refine, each group of scatterers with common f' and f" values is defined via an anomalous_scatterers scope, e.g.:
refinement.refine.anomalous_scatterers {
group {
selection = name BR
f_prime = 0
f_double_prime = 0
refine = *f_prime *f_double_prime
}
}
NOTE: The refinement of the f' and f" values is carried out only if group_anomalous is included under refine.strategy! Otherwise the values are simply used as specified but not refined. So the refinement run with the parameters above included into group_anomalous_1.params: % phenix.refine model.pdb data_anom.hkl group_anomalous_1.params \ strategy=individual_sites+individual_adp+group_anomalous If required, multiple scopes can be specified, one for each unique pair of f' and f" values. These values are assigned to all selected atoms (see below for atom selection details). Often it is possible to start the refinement from zero. If the refinement is not stable, it may be necessary to start from better estimates, or even to fix some values. For example (file group_anomalous_2.params):
refinement.refine.anomalous_scatterers {
group {
selection = name BR
f_prime = -5
f_double_prime = 2
refine = f_prime *f_double_prime
}
}
% phenix.refine model.pdb data_anom.hkl group_anomalous_2.params \
strategy=individual_sites+individual_adp+group_anomalous
Here f' is fixed at -5 (note the missing * in front of f_prime in the refine definition), and the refinement of f" is initialized at 2. The phenix.form_factor_query command is available for obtaining estimates of f' and f" given an element type and a wavelength, e.g.: % phenix.form_factor_query element=Br wavelength=0.8 Information from Sasaki table about Br (Z = 35) at 0.8 A fp: -1.0333 fdp: 2.9928 Run without arguments for usage information: % phenix.form_factor_query Using NCS restraints in refinementphenix.refine can find NCS automatically or use NCS selections defined by the user. Gaps in selected sequences are allowed - a sequence alignment is performed to detect insertions or deletions. We recommend to check the automatically detected or adjusted NCS groups.
Using secondary structure restraintsAt low resolutions it is often beneficial to restrain hydrogen bonding distances in helices, sheets, and nucleic acid base pairs. These can be used with or without explicit hydrogen atoms. Appropriate atom selections will be detected automatically if none are provided by the user, but in most cases careful manual annotation will probably yield better results, especially if the starting model is of low quality. To turn on the additional restraints a single extra parameter is sufficient: % phenix.refine data.hkl model.pdb main.secondary_structure_restraints=True You can also generate starting parameters for secondary structure restraints using a standalone utility: % phenix.secondary_structure_restraints model.pdb This will print a set of parameters suitable for use in phenix.refine, which may be edited to correct errors or add undetected groups. Like other restraints, the hydrogen bond distances have adjustable sigma and target values; these are defined in the hydrogen_bonding scope. The default potential is labeled 'simple', and mimics the covalent bond restraints. The sigma defaults to 0.05 Angstrom; the targets will be different depending on whether explicit hydrogens are used or not (defaults are 1.975A or 2.9A): % phenix.refine data.hkl model.pdb main.secondary_structure_restraints=True \ hydrogen_bonding.distance_ideal_h_o=2.0 \ hydrogen_bonding.simple.sigma=0.04 % phenix.refine data.hkl model.pdb main.secondary_structure_restraints=True \ hydrogen_bonding.distance_ideal_n_o=3.0 \ hydrogen_bonding.simple.slack=0.1 A relatively strict outlier cutoff is applied by default, to prevent improperly restraining incorrectly annotated residues. Any bonds longer than the outlier cutoffs (2.5A for H-O, or 3.5A for N-O) will be be weighted down to zero during refinement (they may contribute later if the bond length decreases). If you are certain of your annotations, you can increase or remove the cutoff:
Using a reference model in refinementphenix.refine can be given a reference model that is used to steer refinement of the working model. This technique is advantageous in cases where the working data set is low resolution, but there is a known related structure solved at higher resolution. The higher resolution reference model is used to generate a set of dihedral restraints that are applied to each matching dihedral in the working model. Reference chains are matched to working chains automatically, and sequences need not match exactly. The default parameters are a good starting point: % phenix.refine data.hkl model.pdb main.reference_model_restraints=True \ reference_model.file=reference.pdb The default sigma value for these reference dihedral restraints is 1.0 Angstrom. To increase the strength of these restraints, select a smaller sigma: % phenix.refine data.hkl model.pdb main.reference_model_restraints=True \ reference_model.file=reference.pdb reference_model.sigma=0.5 To decrease the strength of the restraints, select a larger sigma: % phenix.refine data.hkl model.pdb main.reference_model_restraints=True \ reference_model.file=reference.pdb reference_model.sigma=2.0 The reference restraints have a limit parameter which turns them off when the angle in the working model differs from the reference by an amount greater than limit. The default value is 15 degrees, but may be user-defined: % phenix.refine data.hkl model.pdb main.reference_model_restraints=True \ reference_model.file=reference.pdb reference_model.limit=10 For an optimal set of protein restraints, rotamer outliers in the working model that have rotameric counterparts in the reference model are automatically corrected to the rotamer from the reference model prior to refinement. In practice this step almost always improves the final model, but can be turned off if desired: % phenix.refine data.hkl model.pdb main.reference_model_restraints=True \ reference_model.file=reference.pdb reference_model.fix_outliers=False Selections may also be used with reference_model restraints. Selections are useful in cases where multiple chains in the working model should be restrained to the same reference chain, the model or reference have insertions that change the register, only part of a chain is desirable to restrain, etc. To specify selections, create a reference.params file with selections like:
refinement.reference_model.reference_group {
reference = chain A and resseq 2:119
selection = chain A and resseq 2:119
}
refinement.reference_model.reference_group {
reference = chain A and resseq 130:134
selection = chain A and resseq 120:124
}
refinement.reference_model.reference_group {
reference = chain A
selection = chain B
}
Specify reference.params as an additional input when running phenix.refine: % phenix.refine data.hkl model.pdb main.reference_model_restraints=True \ reference_model.file=reference.pdb reference.params Each selection (both reference and selection entries as above) may only specify one chainID and/or one resseq range. Multiple reference models may also be specified in cases where a working complex has reference structures from different coordinate files. To specify multiple reference model input files, the command line or parameter file should contain: refinement.reference_model.file = reference_A.pdb refinement.reference_model.file = reference_B.pdb Reference chain/model chain pairs are determined automatically, and details are written to the .log file and .eff file. If you want to specify your own matching, include a parameter file that contains:
refinement.reference_model.file = reference_A.pdb
refinement.reference_model.file = reference_B.pdb
refinement.reference_model.reference_group {
reference = chain A
selection = chain A
file_name = reference_A.pdb
}
refinement.reference_model.reference_group {
reference = chain A
selection = chain B
file_name = reference_B.pdb
}
The refinement.reference_model.reference_group.file_name parameter is only required when more than one reference file is used. This parameter allows the reference model restraint generation to disambiguate between reference files that contain chains with the same chainID. Automatic Asn/Gln/His correctionsAsn, Gln, and His residues can often be fit favorably to the data in two orientations, related by a 180 degree rotation. In many cases, however, only one of these orientations is sterically and electrostatically favorable. phenix.refine uses Reduce to identify Asn, Gln, and His residues that should be flipped, and then flips them automatically. % phenix.refine data.hkl model.pdb main.nqh_flips=True Water pickingphenix.refine has very efficient and fully automated protocol for water picking and refinement. One run of phenix.refine is normally necessary to locate waters, refine them, select good ones, add new and refine again, repeating the whole process multiple times. Normally, the default parameter settings are good for most cases: % phenix.refine data.hkl model.pdb ordered_solvent=true This will perform new water picking, analysis of existing waters and refinement of individual coordinates and B-factors for both, macromolecule and waters. Several cycles will be performed allowing sorting out of spurious waters and refinement of well placed ones. Water picking can be combined with all others protocols, like simulated annealing, TLS refinement, etc. Some useful commands are:
Hydrogens in refinementDepending on data type (X-ray or neutron), data quality (resolution, completeness) phenix.refine offers different options for parametrization of hydrogen atoms:
Using the riding model does not add additional refinable parameters, since position of a hydrogen atom H in X-H bond is recalculated from the current position of atom X. Also, H atom inherits the occupancy of X atom and its B-factor. Sometime the B-factor of H atom is the product of B-factor of X atoms and a scale from 1 to 1.5. The riding model should be used to parametrize H atoms at almost all resolutions in X-ray refinement. An exception can be a subatomic resolution ( ~0.7A and higher), where the hydrogen's parameters can be refined individually. Although the contribution of hydrogen atoms to X-ray scattering is weak, the H atoms are still present in real structures irrespective the data quality. Including them as riding model at any resolution makes other model atoms aware of their positions and hence preventing non-physical (bad) contacts at no cost in terms of refinable parameters (= no risk of overfitting). The scattering contribution of Hydrogens by default is always accounted for, however there is a parameter to turn it off: % phenix.refine model.pdb data.hkl hydrogens.contribute_to_f_calc=false If neutron data is used then the parameters of H atoms should always be refined individually, except the cases where data resolution and/or completeness are poor. In that case riding model can be used. If partially deuterated structure is used in refinement then the constrained occupancies of exchangeable H/D sites are refined so they add up to 1. Currently, phenix.refine does not add the H atoms (except a few cases mentioned below), so it is necessary to use a different program from PHENIX suite (ReadySet!) to add H, D or H/D atoms to your structure. Internally ReadySet! uses Reduce program to add hydrogens to macromolecule (protein, DNA/RNA) and it uses its own resources to add hydrogens to ligands or water. Hydrogens are added to their ideal geometrical positions. If a structure contains a ligand unknown to phenix.refine, ReadySet! will create a library CIF file which will include the definitions for all newly added hydrogens. phenix.refine can build H or D atoms for water molecules only. To do so it uses residual density map, mFo-DFc. This option is normally used at relatively high resolution neutron data (~2.0...2.5A and higher) or at subatomic X-ray resolution: % phenix.refine model.pdb data.hkl main.find_and_add_hydrogens=true Although this is not thoroughly studied yet, it seems that hydrogens should not be included into NCS or TLS groups. This is why they are automatically excluded from them. However, if NCS selections are created manually and the structure contains H atoms, it might be a good idea to add and not (element H or element D) to all selection strings. Below are some useful commands:
Refinement using twinned dataphenix.refine can handle the refinement of hemihedrally twinned data (two twin domains). Least square twin refinement can be carried out using the following commands line instructions: % phenix.refine data.hkl model.pdb twin_law="-k,-h,-l" The twin law (in this case -k,-h,-l) can be obtained from phenix.xtriage. If more than a single twin law is possible for the given unit cell and space group, using phenix.twin_map_utils might give clues which twin law is the most likely candidate to be used in refinement. Correcting maps for anisotropy might be useful: % phenix.refine data.hkl model.pdb twin_law="-k,-h,-l" \ detwin.map_types.aniso_correct=true The detwinning mode is auto by default: it will perform algebraic detwinning for twin fraction below 40%, and detwinning using proportionality rules (SHELXL style) for fractions above 40%. An important point to stress is that phenix.refine will only deal properly with twinning that involves two twin domains. Neutron and joint X-ray and neutron refinementRefinement using neutron data requires having H or/and D atoms added to the model. Use ReadySet! program to add all H, D or H/D atoms. See "Hydrogens in refinement" section for details.
Optimizing target weightsphenix.refine uses automatic procedure to determine the weights between X-ray target and stereochemistry or ADP restraints. To optimize these weights (that is to find those resulting in lowest Rfree factors): % phenix.refine data.hkl model.pdb optimize_xyz_weight=true optimize_adp_weight=true where optimize_xyz_weight will turn on the optimization of X-ray/stereochemistry weight and optimize_adp_weight will turn on the optimization of X-ray/ADP weight. Note that this could be very slow since the procedure involves a grid search over an array of weights-candidates. It could be a good idea to run this overnight for a final model tune up. Refinement at high resolution (higher than approx. 1.0 Angstrom)Guidelines for structure refinement at high resolution:
This example illustrates most of the above points: % phenix.refine model_h.pdb data.hkl high_res.params where the file high_res.params contains following lines (for more parameters under each scope look at complete list of parameters):
refinement.main {
number_of_macro_cycles = 5
ordered_solvent=true
}
refinement.refine {
adp {
individual {
isotropic = element H
anisotropic = not element H
}
}
}
refinement.target_weights {
wxc_scale = 0.25
wxu_scale = 0.3
}
refinement {
ordered_solvent {
mode = auto filter_only *every_macro_cycle
new_solvent = isotropic *anisotropic
refine_occupancies = True
}
}
In the example above phenix.refine will perform 5 macro-cycles with ordered solvent update (add/remove) every macro-cycles, all atoms including newly added water will be refined with anisotropic B-factors (except hydrogens), riding model will be used for positional refinement of H atoms, one occupancy and isotropic B-factor will be refined per all hydrogens within a residue, occupancies of waters will be refined as well, the default stereochemistry and ADP restraints weights are scaled down by the factors of 0.25 and 0.3 respectively. If starting model is far enough from the "final" one, more macro-cycles may be required (than 5 used in this example). Examples of frequently used refinement protocols, common problems
Useful optionsChanging the number of refinement cycles and minimizer iterations% phenix.refine data.hkl model.pdb main.number_of_macro_cycles=5 \ main.max_number_of_iterations=20 Parallelizing for multi-core systems% phenix.refine data.hkl model.pdb optimize_xyz_weight=True nproc=4 The nproc parameter instructures phenix.refine to use multiple processors for several highly parallel routines. Currently this applies to the following procedures:
When used with the default settings, nproc will have a minimal effect on overall runtime, but when the optimization grid searches are enabled, a speedup of 4-5x is possible. Values of nproc above 18 are unlikely to yield further speed improvement. Note: this parallelization method is not compatible with OpenMP, and is limited to Mac and Linux systems. (It is, however, available in the Phenix GUI.) Creating R-free flags (if not present in the input reflection files)% phenix.refine data.hkl model.pdb xray_data.r_free_flags.generate=True It is important to understand that reflections selected for test set must be never used in any refinement of any parameters. If the newly selected test reflections were used in refinement before then the corresponding R-free statistics will be wrong. In such case "refinement memory" removal procedure must be applied to recover proper statistics. To change the default maximal number of test flags to be generated and the fraction: % phenix.refine data.hkl model.pdb xray_data.r_free_flags.generate=True \ xray_data.r_free_flags.fraction=0.05 xray_data.r_free_flags.max_free=500 Specify the name for output files% phenix.refine data.hkl model.pdb output.prefix=lysozyme Reflection outputAt the end of refinement a file with Fobs, Fmodel, Fcalc, Fmask, FOM, R-free_flags can be written out (in MTZ format): % phenix.refine data.hkl model.pdb export_final_f_model=mtz Note: Fmodel is the total model structure factor including all scales: Fmodel = scale_k1 * exp(-h*U_overall*ht) * (Fcalc + k_sol * exp(-B_sol*s^2) * Fmask) Setting the resolution range for the refinement% phenix.refine data.hkl model.pdb xray_data.low_resolution=15.0 xray_data.high_resolution=2.0 Bulk solvent correction and anisotropic scalingBy default phenix.refine always starts with bulk solvent modeling and anisotropic scaling. Here is the list of command that may be of use in some cases:
By default phenix.refine adds isotropic component of overall anisotropic scale matrix to atomic B-factors, leaving the trace of overall anisotropic scale matrix equals to zero. This is the reason why one can observe the ADP changed even though the only anisotropic scaling was done and no ADP refinement performed. Default refinement with user specified X-ray target function
Modifying the initial model before refinement startsphenix.refine offers several options to modify input model before refinement starts:
By default, the specified manipulations will be applied to all atoms. However, it is possible to apply them to only selected atoms: % phenix.refine data.hkl model.pdb adp.set_b_iso=25 modify_start_model.selection="chain A" To write out the modified model (without any refinement), add: main.number_of_macro_cycles=0, e.g.: % phenix.refine data.hkl model.pdb adp.set_b_iso=25 \ main.number_of_macro_cycles=0 All the commands listed above plus some more are available from phenix.pdbtools utility which in fact is used internally in phenix.refine to perform these manipulations. For more information on phenix.pdbtools type: % phenix.pdbtools --help Documentation on phenix.pdbtools is also available. Refinement using FFT or direct structure factor calculation algorithm% phenix.refine data.hkl model.pdb \ structure_factors_and_gradients_accuracy.algorithm=fft % phenix.refine data.hkl model.pdb \ structure_factors_and_gradients_accuracy.algorithm=direct Ignoring test (free) flags in refinementSometimes one need to use all reflections ("work" and "test") in the refinement; for example, at very low resolution where each single reflection counts, or at subatomic resolution where the risk of overfitting is very low. In the example below all the reflections are used in the refinement: % phenix.refine data.hkl model.pdb xray_data.r_free_flags.ignore_r_free_flags=true Note: 1) the corresponding statistics (R-factors, ...) will be identical for "work" and "test" sets; 2) it is still necessary to have test flags presented in input reflection file (or automatically generated by phenix.refine). Using phenix.refine to calculate structure factorsThe total structure factor used in phenix.refine nearly in all calculations is defined as: Fmodel = scale_k1 * exp(-h*U_overall*ht) * (Fcalc + k_sol * exp(-B_sol*s^2) * Fmask)
Scattering factorsThere are four choices for the scattering table to be used in phenix.refine:
The default is n_gaussian. To switch to different table: % phenix.refine data.hkl model.pdb main.scattering_table=neutron Suppressing the output of certain filesThe following command will tell phenix,refine to not write .eff, .geo, .def, maps and map coefficients files: % phenix.refine data.hkl model.pdb write_eff_file=false write_geo_file=false \ write_def_file=false write_maps=false write_map_coefficients=false The only output will be: .log and .pdb files. Random seed% phenix.refine data.hkl model.pdb main.random_seed=7112384 The results of certain refinement protocols, such as restrained refinement of coordinates (with SA or LBFGS minimization), are sensitive to the random seed. This is because: 1) for SA the refinement starts with random assignment of velocities to atoms; 2) the X-ray/geometry target weight calculation involves model shaking with some Cartesian dynamics. As result, running such refinement jobs with exactly the same parameters but different random seeds will produce different refinement statistics. The author's experience includes the case where the difference in R-factors was about 2.0% between two SA runs. Also, this opens a possibility to perform multi-start SA refinement to create an ensemble of slightly different models in average but sometimes containing significant variations in certain parts. Electron density mapsBy default phenix.refine outputs two likelihood-weighted maps: 2mFo-DFc and mFo-DFc. These are the map coefficients generated for use in Coot. The user can also choose between likelihood-weighted or regular maps with any specified coefficients, for example: 2mFo-DFc, 2.7mFo-1.3DFc, Fo-Fc, 3Fo-2Fc. Any number of maps can be created. Optionally, the result can be output as binary CCP4 format. The example below illustrates the main options: % phenix.refine data.hkl model.pdb map.params write_maps=true
refinement {
electron_density_maps {
map_coefficients {
mtz_label_amplitudes = 2FOFCWT
mtz_label_phases = PH2FOFCWT
map_type = 2mFo-DFc
}
map_coefficients {
mtz_label_amplitudes = FOFCWT
mtz_label_phases = PHFOFCWT
map_type = mFo-DFc
}
map_coefficients {
mtz_label_amplitudes = 3FO2FCWT
mtz_label_phases = PH3FO2FCWT
map_type = 3Fo-2Fc
}
map {
map_type = 2mFo-DFc
grid_resolution_factor = 1/4.
region = *selection cell
atom_selection = chain A and resseq 1
}
}
}
This will output one file with map coefficients for 2mFo-DFc, mFo-DFc and 3Fo-2Fc maps, and one X-plor formatted file containing 2mFo-DFc map computed around residue 1 in chain A. The map finesse will be (data resolution)*grid_resolution_factor. If atom_selection is set to None or all then map will be computed for all atoms. Refining with anomalous data (or what phenix.refine does with Fobs+ and Fobs-).The way phenix.refine uses Fobs+ and Fobs- is controlled by xray_data.force_anomalous_flag_to_be_equal_to parameter.
Look this documentation to see how to use and refine f' and f''. Rejecting reflections by sigmaReflections can be rejected by sigma cutoff criterion applied to amplitudes Fobs <= sigma_fobs_rejection_criterion * sigma(Fobs): % phenix.refine model.pdb data_anom.hkl xray_data.sigma_fobs_rejection_criterion=2 or/and intensities Iobs <= sigma_iobs_rejection_criterion * sigma(Iobs): % phenix.refine model.pdb data_anom.hkl xray_data.sigma_iobs_rejection_criterion=2 Internally, phenix.refine uses amplitudes. If both sigma_fobs_rejection_criterion and sigma_iobs_rejection_criterion are given as non-zero values, then both criteria will be applied: first to Iobs, then to Fobs (after truncated Iobs got converted to Fobs): % phenix.refine model.pdb data_anom.hkl xray_data.sigma_fobs_rejection_criterion=2 \ xray_data.sigma_iobs_rejection_criterion=2 By default, both sigma_fobs_rejection_criterion and sigma_iobs_rejection_criterion are set to zero (no reflections rejected) and, unless strongly motivated, we encourage to not change these values. If amplitudes provided at input then sigma_fobs_rejection_criterion is ignored. Developer's toolsphenix.refine offers a broad functionality for experimenting that may not be useful in everyday practice but handy for testing ideas. Substitute input Fobs with calculated Fcalc, shake model and refine it
CIF modifications and linksphenix.refine uses the CCP4 monomer library to build geometry restraints (bond, angle, dihedral, chirality and planarity restraints). The CCP4 monomer library comes with a set of "modifications" and "links" which are defined in the file mon_lib_list.cif. Some of these are used automatically when phenix.refine builds the geometry restraints (e.g. the peptide and RNA/DNA chain links). Other links and modifications have to be applied manually, e.g. (cif_modification.params file):
refinement.pdb_interpretation.apply_cif_modification {
data_mod = 5pho
residue_selection = resname GUA and name O5T
}
Here a custom 5pho modification is applied to all GUA residues with an O5T atom. I.e. the modification can be applied to multiple residues with a single apply_cif_modification block. The CIF modification is supplied as a separate file on the phenix.refine command line, e.g. (data_mod_5pho.cif file): data_mod_5pho # loop_ _chem_mod_atom.mod_id _chem_mod_atom.function _chem_mod_atom.atom_id _chem_mod_atom.new_atom_id _chem_mod_atom.new_type_symbol _chem_mod_atom.new_type_energy _chem_mod_atom.new_partial_charge 5pho add . O5T O OH . loop_ _chem_mod_bond.mod_id _chem_mod_bond.function _chem_mod_bond.atom_id_1 _chem_mod_bond.atom_id_2 _chem_mod_bond.new_type _chem_mod_bond.new_value_dist _chem_mod_bond.new_value_dist_esd 5pho add O5T P coval 1.520 0.020 % phenix.refine model_o5t.pdb data.hkl data_mod_5pho.cif cif_modification.params Similarly, a link can be applied like this (cif_link.params file):
refinement.pdb_interpretation.apply_cif_link {
data_link = MAN-THR
residue_selection_1 = chain X and resname MAN and resid 900
residue_selection_2 = chain X and resname THR and resid 42
}
% phenix.refine model.pdb data.hkl cif_link.params
The residue selections for links must select exactly one residue each. The MAN-THR link is pre-defined in mon_lib_list.cif. Custom links can be supplied as additional files on the phenix.refine command line. See mon_lib_list.cif for examples. The full path to this file can be obtained with the command: % phenix.where_mon_lib_list_cif All apply_cif_modification and apply_cif_link definitions will be included into the .def files. I.e. it is not necessary to specify the definitions again if further refinement runs are started with .def files. Note that all LINK, SSBOND, HYDBND, SLTBRG and CISPEP records in the input PDB files are ignored. Definition of custom bonds and anglesMost geometry restraints (bonds, angles, etc.) are generated automatically based on the CCP4 monomer library. Additional custom bond and angle restraints, e.g. between protein and a ligand or ion, can be specified in this way:
refinement.geometry_restraints.edits {
zn_selection = chain X and resname ZN and resid 200 and name ZN
his117_selection = chain X and resname HIS and resid 117 and name NE2
asp130_selection = chain X and resname ASP and resid 130 and name OD1
bond {
action = *add
atom_selection_1 = $zn_selection
atom_selection_2 = $his117_selection
symmetry_operation = None
distance_ideal = 2.1
sigma = 0.02
slack = None
}
bond {
action = *add
atom_selection_1 = $zn_selection
atom_selection_2 = $asp130_selection
symmetry_operation = None
distance_ideal = 2.1
sigma = 0.02
slack = None
}
angle {
action = *add
atom_selection_1 = $his117_selection
atom_selection_2 = $zn_selection
atom_selection_3 = $asp130_selection
angle_ideal = 109.47
sigma = 5
}
}
The atom selections must uniquely select a single atom. Save the geometry_restraints.edits to a file and specify the file name as an additional argument when running phenix.refine for the first time. For example: % phenix.refine model.pdb data.hkl restraints_edits.params The edits will be included into the .def files. I.e. it is not necessary to manually specify them again if further refinement runs are started with .def files. For bonds to symmetry copies, specify the symmetry operation in xyz notation, for example: symmetry_operation = -x-1/2,y-1/2,-z+1/2 To obtain the symmetry_operation, either use Coot (turn on drawing on symmetry copies, then click on the copy and look for the symmetry operation in the status bar), or run this command: iotbx.show_distances your.pdb > all_distances This will produce a potentially long all_distances file, but if you search for sym= there will probably only be a few matches from which it is easy to pick the one you are interested in, based on the pdb atom labels. The bond.slack parameter above can be used to disable a bond restraint within the slack tolerance around distance_ideal. This is useful for hydrogen bond restraints, or when refining with very high-resolution data (e.g. better than 1 A). The bond restraint is activated only if the discrepancy between the model bond distance and distance_ideal is greater than the slack value. The slack is subtracted from the discrepancy. The resulting potential is called a "square-well potential" by some authors. The formula for the contribution to the refinement target function is: weight * delta_slack**2 delta_slack = sign(delta) * max(0, (abs(delta) - slack)) delta = distance_ideal - distance_model weight = 1 / sigma**2 The slack value must be greater than or equal to zero (it can also be None, which is equivalent to zero in this case). Atom selection examplesall All C-alpha atoms (not case sensitive) name ca All atoms with ``H`` in the name (``*`` is a wildcard character) name *H* Atoms names with ``*`` (backslash disables wildcard function) name o2\* name 'O 1' Atom names with primes don't necessarily have to be quoted name o2' Boolean ``and``, ``or`` and ``not`` resname ALA and (name ca or name c or name n or name o) chain a and not altid b resid 120 and icode c and model 2 segid a and element c and charge 2+ and anisou resseq 188 resid is a synonym for resseq: resid 188 Note that if there are several chains containing residue number 188, all of them will be selected. To be more specific and select residue 188 in particular chain: chain A and resid 188 this will select residue 188 only in chain A. Residues 2 through 10 (including 2 and 10) resseq 2:10 resname ALA and backbone resname ALA and sidechain peptide backbone rna backbone or dna backbone water or nucleotide dna and not (phosphate or ribose) within(5, (nucleotide or peptide) backbone) Depositing refined structure with PDBphenix.refine reports a comprehensive statistics in PDB file header of refined model. This statistics consists of two parts: the first (upper, formatted with REMARK record) part is relevant to the current refinement run and contains the information about input data and model files, time stamp, start and final R-factors, refinement statistics from macro-cycle to macro-cycle, etc. The second (lower, formatted with REMARK 3 record) part is abstracted from a particular refinement run (no intermediate statistics, time, no file names, etc.). This part is supposed to go in PDB and the first part should be removed manually. Referencing phenix.refineAfonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr. 68:352-67. PMID: 22505256 Relevant readingBelow is the list of papers either published in connection with phenix.refine or used to implement specific features in phenix.refine:
Feedback, more information
List of all refinement keywords
-------------------------------------------------------------------------------
Legend: black bold - scope names
black - parameter names
red - parameter values
blue - parameter help
blue bold - scope help
Parameter values:
* means selected parameter (where multiple choices are available)
False is No
True is Yes
None means not provided, not predefined, or left up to the program
"%3d" is a Python style formatting descriptor
-------------------------------------------------------------------------------
refinement Scope of parameters for structure refinement with phenix.refine
crystal_symmetry Scope of space group and unit cell parameters
unit_cell= None
space_group= None
input Scope of input file names, labels, processing directions
symmetry_safety_check= *error warning Check for consistency of crystal
symmetry from model and data files
pdb
file_name= None Model file(s) name (PDB)
neutron_data Scope of neutron data and neutron free-R flags
ignore_xn_free_r_mismatch= False
file_name= None
labels= None
high_resolution= None
low_resolution= None
outliers_rejection= True Remove basic wilson outliers , extreme
wilson outliers , and beamstop shadow outliers
french_wilson_scale= True
sigma_fobs_rejection_criterion= None
sigma_iobs_rejection_criterion= None
ignore_all_zeros= True
force_anomalous_flag_to_be_equal_to= None
french_wilson
max_bins= 60 Maximum number of resolution bins
min_bin_size= 40 Minimum number of reflections per bin
r_free_flags
file_name= None This is normally the same as the file containing
Fobs and is usually selected automatically.
label= None
test_flag_value= None This value is usually selected automatically
- do not change unless you really know what
you're doing!
ignore_r_free_flags= False Use all reflections in refinement (work
and test)
disable_suitability_test= False
ignore_pdb_hexdigest= False If True, disables safety check based
on MD5 hexdigests stored in PDB files
produced by previous runs.
generate= False Generate R-free flags (if not available in input
files)
fraction= 0.1
max_free= 2000
lattice_symmetry_max_delta= 5
use_lattice_symmetry= True
use_dataman_shells= False Used to avoid biasing of the test set by
certain types of non-crystallographic
symmetry.
n_shells= 20
xray_data Scope of X-ray data and free-R flags
file_name= None
labels= None
high_resolution= None
low_resolution= None
outliers_rejection= True Remove basic wilson outliers , extreme
wilson outliers , and beamstop shadow outliers
french_wilson_scale= True
sigma_fobs_rejection_criterion= None
sigma_iobs_rejection_criterion= None
ignore_all_zeros= True
force_anomalous_flag_to_be_equal_to= None
french_wilson
max_bins= 60 Maximum number of resolution bins
min_bin_size= 40 Minimum number of reflections per bin
r_free_flags
file_name= None This is normally the same as the file containing
Fobs and is usually selected automatically.
label= None
test_flag_value= None This value is usually selected automatically
- do not change unless you really know what
you're doing!
ignore_r_free_flags= False Use all reflections in refinement (work
and test)
disable_suitability_test= False
ignore_pdb_hexdigest= False If True, disables safety check based
on MD5 hexdigests stored in PDB files
produced by previous runs.
generate= False Generate R-free flags (if not available in input
files)
fraction= 0.1
max_free= 2000
lattice_symmetry_max_delta= 5
use_lattice_symmetry= True
use_dataman_shells= False Used to avoid biasing of the test set by
certain types of non-crystallographic
symmetry.
n_shells= 20
experimental_phases Scope of experimental phase information (HL
coefficients)
file_name= None
labels= None
monomers Scope of monomers information (CIF files)
file_name= None Monomer file(s) name (CIF)
sequence
file_name= None Sequence data in a text file (supported formats
include FASTA, PIR, and raw text). Currently this is only
used by the PHENIX GUI for validation.
output Scope for output files
prefix= None Prefix for all output files
serial= None Serial number for consequtive refinement runs
serial_format= "%03d" Format serial number in output file name
title= None Brief string describing run
write_eff_file= True
write_geo_file= True
write_final_geo_file= False
write_def_file= True
write_model_cif_file= False
write_reflection_cif_file= False
export_final_f_model= mtz cns Write Fobs, Fmodel, various scales and
more to MTZ or CNS file
write_maps= False
write_map_coefficients= True
pickle_fmodel= False Dump final fmodel object into a pickle file.
electron_density_maps Electron density maps calculation parameters
apply_default_maps= None
map_coefficients
map_type= None
format= *mtz phs
mtz_label_amplitudes= None
mtz_label_phases= None
kicked= False
fill_missing_f_obs= False
acentrics_scale= 2.0 Scale terms corresponding to acentric
reflections (residual maps only: k==n)
centrics_pre_scale= 1.0 Centric reflections, k!=n and k*n != 0:
max(k-centrics_pre_scale,0)*Fo-max(n-centrics_pre_
scale,0)*Fc
sharpening= False Apply B-factor sharpening
sharpening_b_factor= None Optional sharpening B-factor value
exclude_free_r_reflections= False Exclude free-R selected reflections
from output map coefficients
isotropize= True
dev
complete_set_up_to_d_min= False
aply_same_incompleteness_to_complete_set_at= randomly low high
map
map_type= None
format= xplor *ccp4
file_name= None
kicked= False
fill_missing_f_obs= False
grid_resolution_factor= 1/4.
scale= *sigma volume
region= *selection cell
atom_selection= None
atom_selection_buffer= 3
acentrics_scale= 2.0 Scale terms corresponding to acentric
reflections (residual maps only: k==n)
centrics_pre_scale= 1.0 Centric reflections, k!=n and k*n != 0:
max(k-centrics_pre_scale,0)*Fo-max(n-centrics_pre_
scale,0)*Fc
sharpening= False Apply B-factor sharpening
sharpening_b_factor= None Optional sharpening B-factor value
exclude_free_r_reflections= False Exclude free-R selected reflections
from map calculation
isotropize= True
refine Scope of refinement flags (=flags defining what to refine) and atom
selections (=atoms to be refined)
strategy= *individual_sites *individual_sites_real_space rigid_body
*individual_adp group_adp tls *occupancies group_anomalous
Atomic parameters to be refined
sites Scope of atom selections for coordinates refinement
individual= None Atom selections for individual atoms
torsion_angles= None Atom selections for Torsion Angle Refinement and
Dynamics
rigid_body= None Atom selections for rigid groups
adp Scope of atom selections for ADP (Atomic Displacement Parameters)
refinement
group_adp_refinement_mode= *one_adp_group_per_residue
two_adp_groups_per_residue group_selection
Select one of three available modes for
group B-factors refinement. For two groups
per residue, the groups will be main-chain
and side-chain atoms. Provide selections
for groups if group_selection is chosen.
group= None One isotropic ADP for group of selected here atoms will
be refined
tls= None Selection(s) for TLS group(s)
individual Scope of atom selections for refinement of individual ADP
isotropic= None Selections for atoms to be refinement with
isotropic ADP
anisotropic= None Selections for atoms to be refinement with
anisotropic ADP
occupancies Scope of atom selections for occupancy refinement
individual= None Selection(s) for individual atoms. None is default
which is to refine the individual occupancies for atoms
in alternative conformations or for atoms with partial
occupancies only.
remove_selection= None Occupancies of selected atoms will not be
refined (even though they might satisfy the default
criteria for occupancy refinement).
constrained_group Selections to define constrained occupancies. If
only one selection is provided then one occupancy
factor per selected atoms will be refined and it
will be constrained between predefined max and min
values.
selection= None Atom selection string.
anomalous_scatterers
group
selection= None
f_prime= 0
f_double_prime= 0
refine= *f_prime *f_double_prime
main Scope for most common and frequently used parameters
bulk_solvent_and_scale= True Do bulk solvent correction and anisotropic
scaling
fix_rotamers= False
flip_peptides= False
nqh_flips= True
simulated_annealing= False Do simulated annealing
simulated_annealing_torsion= False Do simulated annealing in torsion
angle space
ordered_solvent= False Add (or/and remove) and refine ordered solvent
molecules (water)
ncs= False Use restraints NCS in refinement (can be determined
automatically)
ias= False Build and use IAS (interatomic scatterers) model (at
resolutions higher than approx. 0.9 A)
number_of_macro_cycles= 3 Number of macro-cycles to be performed
max_number_of_iterations= 25
use_form_factor_weights= False
tan_u_iso= False Use tan() reparameterization in ADP refinement
(currently disabeled)
use_geometry_restraints= True
secondary_structure_restraints= False Adds distance restraints for
hydrogen bonds involved in secondary
structure. Annotation will be done
automatically if no helix or sheet
records are specified, but this depends
on having a good starting structure.
Nucleic acid base pairs (Watson-Crick
and G-U only) will also be restrained if
present.
hydrogen_bonds= False
reference_model_restraints= False Restrains the dihedral angles to a
high-resolution reference structure to
reduce overfitting at low resolution. You
will need to specify a reference PDB file
(in the input list in the main window) to
use this option.
use_convergence_test= False Determine if refinement converged and stop
then
target= *ml mlhl ml_sad ls Choices for refinement target
min_number_of_test_set_reflections_for_max_likelihood_target= 50 minimum
number of
test
reflections
required
for use of
ML target
max_number_of_resolution_bins= 30
reference_xray_structure= None
show_map_histogram= None
use_experimental_phases= None Use experimental phases if available. If
true, the target function must be set to mlhl ,
and a file containing Hendrickson-Lattman
coefficients must be supplied.
random_seed= 2679941 Ransom seed
scattering_table= wk1995 it1992 *n_gaussian electron neutron Choices of
scattering table for structure factors calculations
use_normalized_geometry_target= True
target_weights_only= False Calculate target weights only and exit
refinement
use_f_model_scaled= False Use Fmodel structure factors multiplied by
overall scale factor scale_k1
max_d_min= 0.25 Highest allowable resolution limit for refinement
fake_f_obs= False Substitute real experimental Fobs with those
calculated from input model (scales and solvent can be
added)
optimize_mask= False Refine mask parameters (solvent_radius and
shrink_truncation_radius)
occupancy_max= 1.0 Maximum allowable occupancy of an atom
occupancy_min= 0.0 Minimum allowable occupancy of an atom
stir= None Stepwise increase of resolution: start refinement at lower
resolution and gradually proceed with higher resolution
rigid_bond_test= False Compute Hirshfeld's rigid bond test value (RBT)
show_residual_map_peaks_and_holes= False Show highest peaks and deepest
holes in residual_map.
fft_vs_direct= False Check accuracy of approximations used in Fcalc
calculations
switch_to_isotropic_high_res_limit= 1.5 If the resolution is lower than
this limit, all atoms selected for
individual ADP refinement and not
participating in TLS groups will be
automatically converted to
isotropic, whether or not ANISOU
records are present in the input PDB
file.
find_and_add_hydrogens= False Find H or D atoms using difference map and
add them to the model. This option should be
used if ultra-high resolution data is available
or when refining againts neutron data.
process_pdb_file_reference= False
correct_special_position_tolerance= 1.0
use_statistical_model_for_missing_atoms= False
set_zero_occupancies_if_in_poor_density= False
nproc= 1 Determines number of processor cores to use in parallel
routines. Currently, this only applies to automatic TLS group
identification.
statistical_model_for_missing_atoms
solvent_content= 0.5
map_type= *2mFo-DFc
resolution_factor= 0.25
probability_mask= True
diff_map_cutoff= 1.5
output_all_masks= False
use_dm_map= False
modify_start_model Scope of parameters to modify initial model before
refinement
selection= None Selection for atoms to be modified
renumber_residues= False Re-number residues
truncate_to_polyala= False Truncate a model to poly-Ala. If True, other
options will be ignored.
remove_alt_confs= False Deletes atoms whose altloc identifier is not
blank or A , and resets the occupancies of the
remaining atoms to 1.0. If True, other options will be
ignored.
set_chemical_element_simple_if_necessary= None Make a simple guess about
what the chemical element is
(based on atom name and the
way how it is formatted) and
write it into output file.
set_seg_id_to_chain_id= False Sets the segID field to the chain ID
(padded with spaces).
clear_seg_id= False Erases the segID field.
remove_first_n_atoms_fraction= None
random_seed= None Random seed
adp Scope of options to modify ADP of selected atoms
atom_selection= None Selection for atoms to be modified. Overrides
parent-level selection.
randomize= False Randomize ADP within a certain range
set_b_iso= None Set ADP of atoms to set_b_iso
convert_to_isotropic= False Convert atoms to isotropic
convert_to_anisotropic= False Convert atoms to anisotropic
shift_b_iso= None Add shift_b_iso value to ADP
scale_adp= None Multiply ADP by scale_adp
sites Scope of options to modify coordinates of selected atoms
atom_selection= None Selection for atoms to be modified. Overrides
parent-level selection.
shake= None Randomize coordinates with mean error value equal to shake
max_rotomer_distortion= None Switch to a rotomer maximally distant
from the current one
min_rotomer_distortion= None Switch to a rotomer minimally distant
from the current one
translate= 0 0 0 Translational shift
rotate= 0 0 0 Rotational shift
euler_angle_convention= *xyz zyz Euler angles convention to be used
for rotation
occupancies Scope of options to modify occupancies of selected atoms
randomize= False Randomize occupancies within a certain range
set= None Set all or selected occupancies to given value
rotate_about_axis
axis= None
angle= None
atom_selection= None
rename_chain_id Rename chains
old_id= None
new_id= None
set_charge
charge_selection= None
charge= None
output Write out PDB file with modified model (file name is defined in
write_modified)
file_name= None Default is the original file name with the file
extension replaced by _modified.pdb .
fake_f_obs Scope of parameters to simulate Fobs
r_free_flags_fraction= None
scattering_table= wk1995 it1992 *n_gaussian neutron Choices of
scattering table for structure factors calculations
fmodel
k_sol= 0.0 Bulk solvent k_sol values
b_sol= 0.0 Bulk solvent b_sol values
b_cart= 0 0 0 0 0 0 Anisotropic scale matrix
scale= 1.0 Overall scale factor
structure_factors_accuracy
algorithm= *fft direct
cos_sin_table= False
grid_resolution_factor= 1/3.
quality_factor= None
u_base= None
b_base= None
wing_cutoff= None
exp_table_one_over_step_size= None
mask
use_asu_masks= True
solvent_radius= 1.11
shrink_truncation_radius= 0.9
grid_step_factor= 4.0 The grid step for the mask calculation is
determined as highest_resolution divided by
grid_step_factor. This is considered as suggested
value and may be adjusted internally based on the
resolution.
verbose= 1
mean_shift_for_mask_update= 0.1 Value of overall model shift in
refinement to updates the mask.
ignore_zero_occupancy_atoms= True Include atoms with zero occupancy
into mask calculation
ignore_hydrogens= True Ignore H or D atoms in mask calculation
n_radial_shells= 1 Number of shells in a radial shell bulk solvent
model
radial_shell_width= 1.3 Radial shell width
hydrogens Scope of parameters for H atoms refinement
refine= individual *riding Choice for refinement: riding model or full
(H is refined as other atoms; useful at very high resolutions
only)
optimize_scattering_contribution= True
contribute_to_f_calc= True Add H contribution to Xray (Fcalc)
calculations
high_resolution_limit_to_include_scattering_from_h= 1.6
real_space_optimize_x_h_orientation= True
xh_bond_distance_deviation_limit= 0.0 Idealize XH bond distances if
deviation from ideal is greater than
xh_bond_distance_deviation_limit
local_real_space_fit_angular_step= 0.5
build
map_type= mFobs-DFmodel Map type to be used to find hydrogens
map_cutoff= 2.0 Map cutoff
angular_step= 3.0 Step in degrees for 6D rigid body search for best
fit
use_sigma_scaled_maps= True Default is sigma scaled map, map in
absolute scale is used otherwise.
resolution_factor= 1./4.
max_number_of_peaks= None
map_next_to_model
min_model_peak_dist= 0.7
max_model_peak_dist= 1.05
min_peak_peak_dist= 1.0
use_hydrogens= False
peak_search
peak_search_level= 1
max_peaks= 0
interpolate= True
min_distance_sym_equiv= None
general_positions_only= False
min_cross_distance= 1.0
min_cubicle_edge= 5
group_b_iso
number_of_macro_cycles= 3
max_number_of_iterations= 25
convergence_test= False
run_finite_differences_test= False
adp
iso
max_number_of_iterations= 25
automatic_randomization_if_all_equal= True
scaling
scale_max= 3.0
scale_min= 10.0
tls
find_automatically= None
one_residue_one_group= None
refine_T= True
refine_L= True
refine_S= True
number_of_macro_cycles= 2
max_number_of_iterations= 25
start_tls_value= None
run_finite_differences_test= False
eps= 1.e-6
min_tls_group_size= 5 min number of atoms allowed per TLS group
adp_restraints
iso
use_u_local_only= False
sphere_radius= 5.0
distance_power= 1.69
average_power= 1.03
wilson_b_weight_auto= False
wilson_b_weight= None
plain_pairs_radius= 5.0
refine_ap_and_dp= False
group_occupancy
number_of_macro_cycles= 3
max_number_of_iterations= 25
convergence_test= False
run_finite_differences_test= False
group_anomalous
number_of_minimizer_cycles= 3
lbfgs_max_iterations= 20
number_of_finite_difference_tests= 0
rigid_body Scope of parameters for rigid body refinement
mode= *first_macro_cycle_only every_macro_cycle Defines how many times
the rigid body refinement is performed during refinement run.
first_macro_cycle_only to run only once at first macrocycle,
every_macro_cycle to do rigid body refinement
main.number_of_macro_cycles times
target= ls_wunit_k1 ml *auto Rigid body refinement target function:
least-squares or maximum-likelihood
target_auto_switch_resolution= 6.0 Used if target=auto, use optimal
target for given working resolution.
disable_final_r_factor_check= False If True, the R-factor check after
refinement will not revert to the previous
model, even if the R-factors have
increased.
refine_rotation= True Only rotation is refined (translation is fixed).
refine_translation= True Only translation is refined (rotation is fixed).
max_iterations= 25 Number of LBFGS minimization iterations
bulk_solvent_and_scale= True Bulk-solvent and scaling within rigid body
refinement (needed since large rigid body shifts
invalidate the mask).
euler_angle_convention= *xyz zyz Euler angles convention
lbfgs_line_search_max_function_evaluations= 10
min_number_of_reflections= 200 Number of reflections that defines the
first lowest resolution zone for the
multiple_zones protocol. If very large
displacements are expected, decreasing this
parameter to 100 may lead to a larger
convergence radius.
multi_body_factor= 1
zone_exponent= 3.0
high_resolution= 3.0 High resolution cutoff (used for rigid body
refinement only)
max_low_high_res_limit= None Maximum value for high resolution cutoff
for the first lowest resolution zone
number_of_zones= 5 Number of resolution zones for MZ protocol
ncs
find_automatically= True If enabled, Phenix will ignore existing
restraint groups and attempt to define appropriate
selections by comparing chains. This only applies to
global NCS restraints - if torsion restraints are
used, the restraint groups will always be defined
automatically unless the user provides custom
selections.
type= *torsion cartesian
coordinate_sigma= None
restrain_b_factors= False If enabled, b-factors will be restrained for
NCS-related atoms. Otherwise, atomic b-factors will
be refined independently, and b_factor_weight will
be set to 0.0
b_factor_weight= None
excessive_distance_limit= 1.5
special_position_warnings_only= False
simple_ncs_from_pdb
pdb_in= None Input PDB file to be used to identify ncs
temp_dir= "" temporary directory (ncs_domain_pdb will be written
there)
min_length= 10 minimum number of matching residues in a segment
njump= 1 Take every njumpth residue instead of each 1
njump_recursion= 10 Take every njump_recursion residue instead of
each 1 on recursive call
min_length_recursion= 50 minimum number of matching residues in a
segment for recursive call
min_percent= 95. min percent identity of matching residues
max_rmsd= 2. max rmsd of 2 chains. If 0, then only search for domains
quick= True If quick is set and all chains match, just look for 1 NCS
group
max_rmsd_user= 3. max rmsd of chains suggested by user (i.e., if
called from phenix.refine with suggested ncs groups)
maximize_size_of_groups= True You can request that the scoring be set
up to maximize the number of members in NCS
groups (maximize_size_of_groups=True) or
that scoring is set up to maximize the
length of the matching segments in the NCS
group (maximize_size_of_groups=False)
require_equal_start_match= True You can require that all matching
segments start at the same relative
residue number for all members of an NCS
group, trimming the matching region as
necessary. This is required if residue
numbers in different chains are not the
same, but not otherwise
ncs_domain_pdb_stem= None NCS domains will be written to
ncs_domain_pdb_stem+"group_"+nn
write_ncs_domain_pdb= False You can write out PDB files representing
NCS domains for density modification if you
want
verbose= False Verbose output
raise_sorry= False Raise sorry if problems
debug= False Debugging output
dry_run= False Just read in and check parameter names
domain_finding_parameters
find_invariant_domains= True Find the parts of a set of chains
that follow NCS
initial_rms= 0.5 Guess of RMS among chains
match_radius= 2.0 Keep atoms that are within match_radius of
NCS-related atoms
similarity_threshold= 0.75 Threshold for similarity between
segments
smooth_length= 0 two segments separated by smooth_length or less
get connected
min_contig_length= 3 segments < min_contig_length rejected
min_fraction_domain= 0.2 domain must be this fraction of a chain
max_rmsd_domain= 2. max rmsd of domains
restraint_group
reference= None
selection= None
coordinate_sigma= 0.05
b_factor_weight= 10
torsion
sigma= 3.0
limit= 15.0
slack= 0.0
similarity= .80
fix_outliers= Auto
check_rotamer_consistency= Auto Check for rotamer differences between
NCS matched sidechains and search for best
fit amongst candidate rotamers
target_damping= False
damping_limit= 10.0
verbose= True
restraint_group
selection= None
b_factor_weight= 10
coordinate_sigma= 0.5
modify_f_obs
remove= random strong weak strong_and_weak low other
remove_fraction= 0.1
fill_mode= fobs_mean_mixed_with_dfmodel random fobs_mean *dfmodel
pdb_interpretation
cdl= False Use Conformation Dependent Library (CDL) for geometry
minimization restraints
correct_hydrogens= False
link_distance_cutoff= 3
disulfide_distance_cutoff= 3
peptide_nucleotide_distance_cutoff= 3
dihedral_function_type= *determined_by_sign_of_periodicity
all_sinusoidal all_harmonic
chir_volume_esd= 0.2
max_reasonable_bond_distance= 50.0
nonbonded_distance_cutoff= None
default_vdw_distance= 1
min_vdw_distance= 1
nonbonded_buffer= 1 **EXPERIMENTAL, developers only**
nonbonded_weight= None Weighting of nonbonded restraints term when
hydrogen atoms are omitted. When explicit hydrogens
are present, the weight will be set to 16, which was
the default in previous versions.
const_shrink_donor_acceptor= 0.6 **EXPERIMENTAL, developers only**
vdw_1_4_factor= 0.8
min_distance_sym_equiv= 0.5
custom_nonbonded_symmetry_exclusions= None
translate_cns_dna_rna_residue_names= None
proceed_with_excessive_length_bonds= False
stop_for_unknowns= True Stop if any nonbonded parameters are unknown.
altloc_weighting
weight= False
bonds= True
angles= True
factor= 1
sqrt= True
apply_cif_modification
data_mod= None
residue_selection= None
apply_cif_link
data_link= None
residue_selection_1= None
residue_selection_2= None
peptide_link
ramachandran_restraints= False Restrains peptide backbone to fall
within allowed regions of Ramachandran plot.
Although it does not eliminate outliers, it
can significantly improve the percent
favored and percent outliers at low
resolution. Probably not useful (and maybe
even harmful) at resolutions much higher
than 3.5A.
cis_threshold= 45
discard_omega= False
discard_psi_phi= True
omega_esd_override_value= None
rama_weight= 1.0
scale_allowed= 1.0
rama_potential= *oldfield emsley
rama_selection= None
rama_exclude_sec_str= False
oldfield
esd= 10.0
weight_scale= 1.0
dist_weight_max= 10.0
weight= None
rna_sugar_pucker_analysis
bond_min_distance= 1.2
bond_max_distance= 1.8
epsilon_range_min= 155.0
epsilon_range_max= 310.0
delta_range_2p_min= 129.0
delta_range_2p_max= 162.0
delta_range_3p_min= 65.0
delta_range_3p_max= 104.0
p_distance_c1p_outbound_line_2p_max= 2.9
o3p_distance_c1p_outbound_line_2p_max= 2.4
bond_detection_distance_tolerance= 0.5
show_histogram_slots
bond_lengths= 5
nonbonded_interaction_distances= 5
bond_angle_deviations_from_ideal= 5
dihedral_angle_deviations_from_ideal= 5
chiral_volume_deviations_from_ideal= 5
show_max_items
not_linked= 5
bond_restraints_sorted_by_residual= 5
nonbonded_interactions_sorted_by_model_distance= 5
bond_angle_restraints_sorted_by_residual= 5
dihedral_angle_restraints_sorted_by_residual= 3
chirality_restraints_sorted_by_residual= 3
planarity_restraints_sorted_by_residual= 3
residues_with_excluded_nonbonded_symmetry_interactions= 12
clash_guard
nonbonded_distance_threshold= 0.5
max_number_of_distances_below_threshold= 100
max_fraction_of_distances_below_threshold= 0.1
geometry_restraints
edits
excessive_bond_distance_limit= 10
bond
action= *add delete change
atom_selection_1= None
atom_selection_2= None
symmetry_operation= None The bond is between atom_1 and
symmetry_operation * atom_2, with atom_1 and
atom_2 given in fractional coordinates.
Example: symmetry_operation = -x-1,-y,z
distance_ideal= None
sigma= None
slack= None
angle
action= *add delete change
atom_selection_1= None
atom_selection_2= None
atom_selection_3= None
angle_ideal= None
sigma= None
planarity
action= *add delete change
atom_selection= None
sigma= None
scale_restraints Apply a scale factor to restraints for specific atom
selections, to tighten geometry without changing the
overall scale of the geometry target.
atom_selection= None
scale= 1.0
apply_to= *bond *angle *dihedral *chirality
geometry_restraints
remove
angles= None
dihedrals= None
chiralities= None
planarities= None
ordered_solvent
low_resolution= 2.8 Low resolution limit for water picking (at lower
resolution water will not be picked even if requessted)
mode= *auto filter_only every_macro_cycle Choices for water picking
strategy: auto - start water picking after ferst few macro-cycles,
filter_only - remove water only, every_macro_cycle - do water
update every macro-cycle
n_cycles= 1
output_residue_name= HOH
output_chain_id= S
output_atom_name= O
scattering_type= O Defines scattering factors for newly added waters
primary_map_type= mFobs-DFmodel
primary_map_cutoff= 3.0
h_bond_min_mac= 1.8
h_bond_min_sol= 1.8
h_bond_max= 3.2
new_solvent= *isotropic anisotropic Based on the choice, added solvent
will have isotropic or anisotropic b-factors
refine_adp= True Refine ADP for newly placed solvent.
b_iso_min= 1.0 Minimum B-factor value, waters with smaller value will be
rejected
b_iso_max= 80.0 Maximum B-factor value, waters with bigger value will be
rejected
anisotropy_min= 0.1 For solvent refined as anisotropic: remove is less
than this value
b_iso= None Initial B-factor value for newly added water
refine_occupancies= False Refine solvent occupancies.
occupancy_min= 0.1 Minimum occupancy value, waters with smaller value
will be rejected
occupancy_max= 1.0 Maximum occupancy value, waters with bigger value
will be rejected
occupancy= 1.0 Initial occupancy value for newly added water
filter_at_start= True
ignore_final_filtering_step= False
correct_drifted_waters= True
use_kick_maps= False Use Dusan's Turk kick maps for peak picking
secondary_map_and_map_cc_filter
cc_map_1_type= "Fc"
cc_map_2_type= 2mFo-DFmodel
atom_radius= 1.5 Atomic radius for map CC calculation. Determined
automatically if if None is given
hydrogen_atom_radius= 1.0 Atomic radius for map CC calculation for H
or D.
number_of_grid_points= 50 Requesteed number of grid points to be used
in CC calculation. Together with atom_radius
it will define the grid step.
set_cc_to_zero_if_n_grid_points_less_than= 10 Return zero CC if
number of grid nodes is
less than defined above
poor_cc_threshold= 0.7 Ad hoc definition of poor map CC
poor_map_value_threshold= 1.0 Ad hoc value (less than) defining a
week density (in sigma) for maps involved
in map CC calculation
crystal_symmetry
unit_cell= None
space_group= None
kick_map parameters for kick maps
kick_size= 0.5
number_of_kicks= 100
peak_search
use_sigma_scaled_maps= True Default is sigma scaled map, map in absolute
scale is used otherwise.
resolution_factor= 1./4.
max_number_of_peaks= None
map_next_to_model
min_model_peak_dist= 1.8
max_model_peak_dist= 6.0
min_peak_peak_dist= 1.8
use_hydrogens= False
peak_search
peak_search_level= 1
max_peaks= 0
interpolate= True
min_distance_sym_equiv= None
general_positions_only= False
min_cross_distance= 1.8
min_cubicle_edge= 5
bulk_solvent_and_scale
mode= slow *fast
bulk_solvent= True
anisotropic_scaling= True
k_sol_b_sol_grid_search= True
minimization_k_sol_b_sol= True
minimization_b_cart= True
target= ls_wunit_k1 *ml
symmetry_constraints_on_b_cart= True
k_sol_max= 0.6
k_sol_min= 0.0
b_sol_max= 300.0
b_sol_min= 0.0
k_sol_grid_search_max= 0.6
k_sol_grid_search_min= 0.0
b_sol_grid_search_max= 80.0
b_sol_grid_search_min= 20.0
k_sol_step= 0.2
b_sol_step= 20.0
number_of_macro_cycles= 2
max_iterations= 25
min_iterations= 25
fix_k_sol= None
fix_b_sol= None
fix_b_cart
b11= None
b22= None
b33= None
b12= None
b13= None
b23= None
alpha_beta
free_reflections_per_bin= 140
number_of_macromolecule_atoms_absent= 225
n_atoms_included= 0
bf_atoms_absent= 15.0
final_error= 0.0
absent_atom_type= "O"
method= *est calc
estimation_algorithm= *analytical iterative
verbose= -1
interpolation= True
fix_scale_for_calc_option= None
number_of_waters_absent= 613
sigmaa_estimator
kernel_width_free_reflections= 100
kernel_on_chebyshev_nodes= True
number_of_sampling_points= 20
number_of_chebyshev_terms= 10
use_sampling_sum_weights= True
mask
use_asu_masks= True
solvent_radius= 1.11
shrink_truncation_radius= 0.9
grid_step_factor= 4.0 The grid step for the mask calculation is
determined as highest_resolution divided by
grid_step_factor. This is considered as suggested
value and may be adjusted internally based on the
resolution.
verbose= 1
mean_shift_for_mask_update= 0.1 Value of overall model shift in
refinement to updates the mask.
ignore_zero_occupancy_atoms= True Include atoms with zero occupancy into
mask calculation
ignore_hydrogens= True Ignore H or D atoms in mask calculation
n_radial_shells= 1 Number of shells in a radial shell bulk solvent model
radial_shell_width= 1.3 Radial shell width
tardy Under development
mode= every_macro_cycle *second_and_before_last once first first_half
xray_weight_factor= 10
start_temperature_kelvin= 2500
final_temperature_kelvin= 300
velocity_scaling= True
temperature_cap_factor= 1.5
excessive_temperature_factor= 5
number_of_cooling_steps= 500
number_of_time_steps= 1
time_step_pico_seconds= 0.001
temperature_degrees_of_freedom= *cartesian constrained
minimization_max_iterations= 0
omit_bonds_with_slack_greater_than= 0
constrain_dihedrals_with_sigma_less_than= 10
near_singular_hinges_angular_tolerance_deg= 5
emulate_cartesian= False
trajectory_directory= None
prolsq_repulsion_function_changes energy(delta) =
c_rep*(max(0,(k_rep*vdw_distance)**irex
p-delta**irexp))**rexp
c_rep= None Usual value: 16
k_rep= 0.75 Usual value: 1. Smaller values reduce the distance
threshold at which the repulsive force becomes active.
irexp= None Usual value: 1
rexp= None Usual value: 4
cartesian_dynamics
temperature= 300
number_of_steps= 200
time_step= 0.0005
initial_velocities_zero_fraction= 0
n_print= 100
verbose= -1
simulated_annealing
start_temperature= 5000
final_temperature= 300
cool_rate= 100
number_of_steps= 50
time_step= 0.0005
initial_velocities_zero_fraction= 0
n_print= 100
update_grads_shift= 0.3
refine_sites= True
refine_adp= False
max_number_of_iterations= 25
mode= every_macro_cycle *second_and_before_last once first first_half
verbose= -1
interleaved_minimization
number_of_iterations= 0
time_step_factor= 10
restraints= *bonds *angles
target_weights
optimize_xyz_weight= False
optimize_adp_weight= False
wxc_scale= 0.5
wxu_scale= 1.0
wc= 1.0
wu= 1.0
fix_wxc= None
fix_wxu= None
shake_sites= True
shake_adp= 10.0
regularize_ncycles= 50
verbose= 1
wnc_scale= 0.5
wnu_scale= 1.0
rmsd_cutoff_for_gradient_filtering= 3.0
force_optimize_weights= False
weight_selection_criteria
bonds_rmsd= None
angles_rmsd= None
r_free_minus_r_work= None
r_free_range_width= None
mean_diff_b_iso_bonded_fraction= None
min_diff_b_iso_bonded= None
ias
b_iso_max= 100.0
occupancy_min= -1.0
occupancy_max= 1.5
ias_b_iso_max= 100.0
ias_b_iso_min= 0.0
ias_occupancy_min= 0.01
ias_occupancy_max= 3.0
initial_ias_occupancy= 1.0
build_ias_types= L R B BH
ring_atoms= None
use_map= True
build_only= False
file_prefix= None
lone_pair
atom_x= CA
atom_xo= C
atom_o= O
peak_search_map
map_type= *Fobs-Fmodel mFobs-DFmodel
grid_step= 0.25
scaling= *volume sigma
ls_target_names
target_name= *ls_wunit_k1 ls_wunit_k2 ls_wunit_kunit ls_wunit_k1_fixed
ls_wunit_k1ask3_fixed ls_wexp_k1 ls_wexp_k2 ls_wexp_kunit
ls_wff_k1 ls_wff_k2 ls_wff_kunit ls_wff_k1_fixed
ls_wff_k1ask3_fixed lsm_kunit lsm_k1 lsm_k2 lsm_k1_fixed
lsm_k1ask3_fixed
twinning
twin_law= None
twin_target= *twin_lsq_f
detwin
mode= algebraic proportional *auto
map_types
twofofc= *two_m_dtfo_d_fc two_dtfo_fc
fofc= *m_dtfo_d_fc gradient m_gradient
aniso_correct= False
structure_factors_and_gradients_accuracy
algorithm= *fft direct
cos_sin_table= False
grid_resolution_factor= 1/3.
quality_factor= None
u_base= None
b_base= None
wing_cutoff= None
exp_table_one_over_step_size= None
r_free_flags
fraction= 0.1
max_free= 2000
lattice_symmetry_max_delta= 5.0 Tolerance used in the determination of
the highest lattice symmetry. Can be thought
of as angle between lattice vectors that
should line up perfectly if the symmetry is
ideal. A typical value is 3 degrees.
use_lattice_symmetry= True When generating Rfree flags, do so in the
asymmetric unit of the highest lattice symmetry.
The result is an Rfree set suitable for twin
refinement.
real_space_refinement
mode= simple diff_map *lockit Real space refinement method (diff_map is
much slower but might have larger convergence radius especially at
low resolution)
target_map_name= 2mFo-DFc Target map type to refine against
model_map_name= Fc Current model map type (used only if diff_map is
selected)
grid_resolution_factor= 1./4 Defines coarseness of the map
target_weights= value grid_search *gradients_ratio Method for relative
target weight determination
real_space_target_weight= None Weight for the real-space target (used
only if diff_map is selected)
restraints_target_weight= None Weight for the restraints target (used
only if diff_map is selected)
number_of_cycles= 1 Number of minimization cycles
rmsd_max_bonds= 0.07 Refinement result is ignored if this max allowable
deviation exceeded
rmsd_max_angles= 5.0 Refinement result is ignored if this max allowable
deviation exceeded
grid_search_scales= 0.25 0.5 0.75 1.0 1.25 1.5 1.75 2.0 2.5 3.0 Defines
the range of values for the weight grid search
verbose= 1 All output is supressed if it is negative
lockit_parameters
lbfgs_max_iterations= 500
finishing_geometry_minimization
cycles_max= 100
first_weight_scale= 0.1
cycle_weight_multiplier= 1.0
superpose_cycle_end_with_cycle_start= False
dihedral_restraints= False
output_file= Auto
real_space_target_weights
first_sample= 10
sampling_step= 30
number_of_samples= 10
bond_rmsd_target= 0.03
worst_acceptable_bond_rmsd
pool_size= 10
max_pool_average= 0.1
fit_side_chains
number_of_macro_cycles= 1
real_space_refine_overall= False
validate_change= True
exclude_hydrogens= True
filter_residual_map_value= 2.0
filter_2fofc_map= None
target_map= 2mFo-DFc
residual_map= mFo-DFc
model_map= Fc
exclude_free_r_reflections= False
use_dihedral_restraints= False
ignore_water_when_move_sidechains= True
residue_iteration
poor_cc_threshold= 0.9
real_space_refine_rotamer= True
real_space_refine_max_iterations= 25
real_space_refine_target_weight= 100.
use_rotamer_iterator= True
torsion_grid_search= True
ignore_alt_conformers= True
torsion_search
min_angle_between_solutions= 5
range_start= -40
range_stop= 40
step= 2
flip_peptides
number_of_macro_cycles= 1
real_space_refine_overall= False
validate_change= True
exclude_hydrogens= True
filter_residual_map_value= 2.0
filter_2fofc_map= None
target_map= 2mFo-DFc
residual_map= mFo-DFc
model_map= Fc
exclude_free_r_reflections= False
ignore_water_when_flipping= True
skip_approximate_helices= True
residue_iteration
poor_cc_threshold= 0.8
real_space_refine_peptide= True
real_space_refine_window= 1
real_space_refine_max_iterations= 25
real_space_refine_target_weight= 100.
torsion_grid_search= True
ignore_alt_conformers= True
torsion_search
min_angle_between_solutions= 5
range_start= -40
range_stop= 40
step= 2
secondary_structure
input
file_name= None
use_hydrogens= True
include_helices= True
include_sheets= True
find_automatically= None
helices_from_phi_psi= False
force_nucleic_acids= False This will ignore the automatic chain type
detection and run the base pair detection using
PROBE even if no nucleic acids are found. Useful
for tRNAs which have a large number of modified
bases.
h_bond_restraints
verbose= False
substitute_n_for_h= None
restrain_helices= True
alpha_only= False
restrain_sheets= True
restrain_base_pairs= True
remove_outliers= None
distance_ideal_n_o= 2.9
distance_cut_n_o= 3.5
distance_ideal_h_o= 1.975
distance_cut_h_o= 2.5
sigma= 0.05
slack= 0.0
top_out= False
helix
selection= None
helix_type= *alpha pi 3_10 unknown Type of helix, defaults to alpha.
Only alpha, pi, and 3_10 helices are used for
hydrogen-bond restraints.
restraint_sigma= None
restraint_slack= None
backbone_only= False Only applies to rigid-body groupings, and not
H-bond restraints which are already backbone-only.
sheet
first_strand= None
restraint_sigma= None
restraint_slack= None
backbone_only= False Only applies to rigid-body groupings, and not
H-bond restraints which are already backbone-only.
strand
selection= None
sense= parallel antiparallel *unknown
bond_start_current= None
bond_start_previous= None
nucleic_acids
sigma= None Defaults to global setting
slack= None Defaults to global setting
use_db_values= True
base_pair
base1= None
base2= None
saenger_class= None reference
leontis_westhof_class= *Auto wwt reference
hydrogen_bonding
restraint_type= *Auto simple lennard_jones implicit
include_side_chains= True
optimize_hbonds= False
optimize_hbonds_thorough= False
optimize_mode= *first last every_macro_cycle
restraints_weight= 1.0
falloff_distance= 0.05
exclude_nonbonded= True
distance_ideal_h_o= 1.975
distance_cut_h_o= 2.5
distance_ideal_n_o= 2.9
distance_cut_n_o= 3.5
implicit Based on H-bond potential for CNS by Chapman lab
theta_high= 155
theta_low= 115
theta_cut= 90
explicit Similar to Rosetta H-bond energy (Kortemme & Baker)
theta_ideal= 180
theta_sigma= 5
psi_ideal= 155
psi_sigma= 5
relative_weights= 1.0 1.0 1.0
lennard_jones
potential= *4_6 6_12
simple Pseudo-bond restraints
sigma= 0.05
slack= 0.0
reference_model
use_distance_based_target= False
file= None
use_starting_model_as_reference= False
sigma= 1.0
limit= 15.0
slack= 0.0
hydrogens= False
main_chain= True
side_chain= True
fix_outliers= True
strict_rotamer_matching= False
auto_shutoff_for_ncs= False
SSM_alignment= True
similarity= .80
secondary_structure_only= False
edits
excessive_bond_distance_limit= 10
bond
action= *add delete change
atom_selection_1= None
atom_selection_2= None
symmetry_operation= None The bond is between atom_1 and
symmetry_operation * atom_2, with atom_1 and
atom_2 given in fractional coordinates.
Example: symmetry_operation = -x-1,-y,z
distance_ideal= None
sigma= None
slack= None
angle
action= *add delete change
atom_selection_1= None
atom_selection_2= None
atom_selection_3= None
angle_ideal= None
sigma= None
planarity
action= *add delete change
atom_selection= None
sigma= None
scale_restraints Apply a scale factor to restraints for specific atom
selections, to tighten geometry without changing the
overall scale of the geometry target.
atom_selection= None
scale= 1.0
apply_to= *bond *angle *dihedral *chirality
remove
angles= None
dihedrals= None
chiralities= None
planarities= None
reference_group
reference= None
selection= None
file_name= None this is to used internally to disambiguate cases
where multiple reference models contain the same chain ID.
This normally does not need to be set by the user
gui Miscellaneous parameters for phenix.refine GUI
base_output_dir= None
tmp_dir= None
send_notification= False
notify_email= None
add_hydrogens= False Runs phenix.ready_set to add hydrogens prior to
refinement.
skip_rsr= False
skip_kinemage= False
phil_file= None
ready_set_hydrogens
add_h_to_water= False
add_d_to_water= False
neutron_exchange_hydrogens= False Add deuteriums in exchangeable sites
perdeuterate= False Add deuteriums in all possible sites
| |