| Python-based Hierarchical ENvironment for Integrated Xtallography |
| Documentation Home |
phenix.superpose_maps
Authors
Guidelines for usephenix.superpose_maps is a utility for transforming two maps to the same frame of reference, allowing direct comparison of molecular features in electron density. The name is slightly misleading, because the program actually performs a superposition of PDB files, then reorients the associated maps to follow them. This is usually done for publication purposes, to compare electron density for the same feature(s) in different crystal forms. The examples shown here are structures of the protein kinase PKA, one in space group P4(2) (PDB ID 1syk), one in space group P2(1)2(1)2(1) (PDB ID 3fhi). (These are included in the PHENIX distribution as part of the "pka-compare" example.) We recommend running this program from the GUI, and this document covers the GUI version; however, the command-line program should work equally well, and parameters are listed below. The program can be started from the main GUI by clicking "Superpose maps" in the "Maps" category. A PDB file and an MTZ file containing map coefficients are required for each structure. Map column labels will be extracted automatically and added to the drop-down menus. 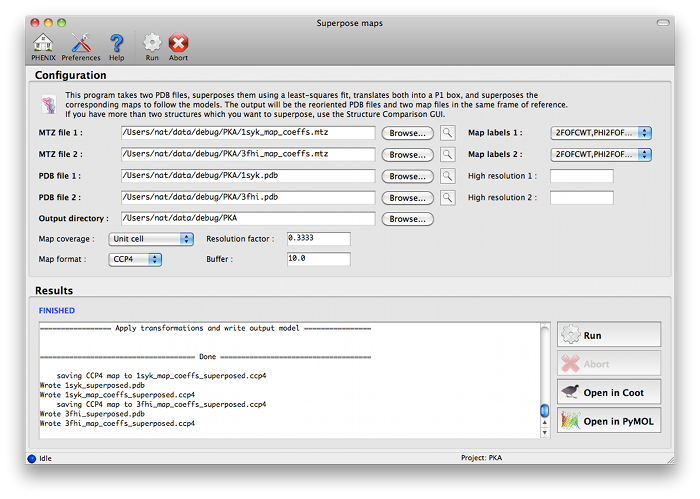 The program will create a new directory named "SuperposeMaps_X", where X is the job ID (this applies to the GUI version only). Output is in either CCP4 or XPLOR map format (we recommend CCP4 maps because of the reduced file size). These can be automatically loaded into Coot or PyMOL once the program is finished running. 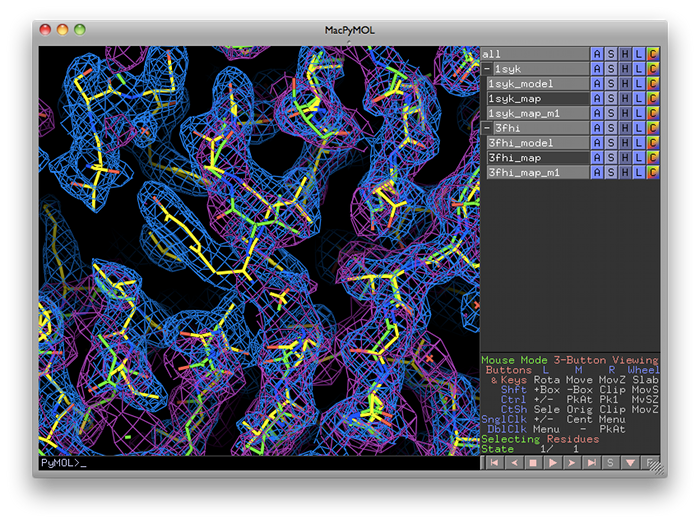 Caveats and known limitations
References
List of all superpose_maps keywords
-------------------------------------------------------------------------------
Legend: black bold - scope names
black - parameter names
red - parameter values
blue - parameter help
blue bold - scope help
Parameter values:
* means selected parameter (where multiple choices are available)
False is No
True is Yes
None means not provided, not predefined, or left up to the program
"%3d" is a Python style formatting descriptor
-------------------------------------------------------------------------------
verbose= False
dry_run= False
superpose_maps
input_files
map_1= None Map coefficients in MTZ format
pdb_1= None PDB file associated with map_1
map_2= None Map coefficients in MTZ format
pdb_2= None
labels_1= None MTZ column labels for map
labels_2= None MTZ column labels for map
d_min_1= None High resolution cutoff for map_1
d_min_2= None High resolution cutoff for map_2
output
output_dir= None
region= *cell selection Region to cover
resolution_factor= 0.3333 Map grid spacing (multiplied times resolution)
map_format= *ccp4 xplor
buffer= 10.0 Padding around molecule
job_title= None
misc
is_difference_map= False
superpose_pdbs
selection_fixed= None Selection of the target atoms to fit to (optional)
selection_moving= None Selection of the atoms that will be fit to
selection_fixed (optional)
selection_default_moving= pepnames and (name ca or name n or name c) and
'altloc " "' Select protein atoms that will be
used in superposition after sequence alignment
selection_default_fixed= pepnames and (name ca or name n or name c) and
'altloc " "' Select protein atoms that will be
used in superposition after sequence alignment
reciprocal_matching= False try both orientations for homodimers
input
pdb_file_name_fixed= None Name of PDB file with model to fit to
pdb_file_name_moving= None Name of PDB file with model that will be
fit to pdb_file_name_fixed
output
file_name= None Name of PDB file with model that best fits to
pdb_file_name_fixed
job_title= None Job title in PHENIX GUI, not used on command line
alignment Set of parameters for sequence alignment. Defaults are good
for most of cases
alignment_style= local *global
gap_opening_penalty= 1
gap_extension_penalty= 1
similarity_matrix= blosum50 dayhoff *identity
| |