refine occupancies for MET/MSE double conformations

Hi there - I was pleased to read in the manual: if no selections are provided (refine.occupancies.individual=None and refine.occupancies.group=None) then phenix.refine by default will refine individual occupancies for atoms that have partial occupancy values in input PDB file (except zeros) and it will perform group constrained occupancy refinement for all atoms in alternative conformations. Occupancies of all other atoms will be fixed. If selections are provided (refine.occupancies.individual=atom_selection or refine.occupancies.group=atom_selection) then the selected atoms will be added to the list of atoms defined above (atoms with partial occupancies or those in alternative conformations). See this document for examples of atom_selection syntax. However, I want to refine my MET residues in double conformations, one as MET (AMET) and the other one as MSE (BMSE) with each 0.5 occupancy to start with. When I try that I get: == ----------Initial model statistics (before refinement)---------- ... |-Occupancies statistics------------------------------------------------------| Sorry: There are atoms with negative occupancies. Check input PDB file. === The PDB file has 0.5 and 1.0 occupancies only. I am also using: == refinement.refine.strategy=tls+individual_sites+individual_adp +group_anomalous refinement.refine.adp.tls="chain A" refinement.refine.adp.tls="chain B" refinement.main.number_of_macro_cycles=4 refinement.simulated_annealing.start_temperature=5000 refinement.refine.adp.individual.anisotropic =" chain C" refinement .input .xray_data .labels=I_Mystery(+),SIGI_Mystery(+),I_Mystery(-),SIGI_Mystery(-),merged refinement.refine.anomalous_scatterers { group { selection = "name ZN " f_prime = -0.3 f_double_prime = 2.5 refine = *f_prime *f_double_prime } group { selection = "name SE " f_prime = -5.2 f_double_prime = 3.8 refine = *f_prime *f_double_prime } } == Any hints on how to proceed ? Should I define groups for the occupancy refinement explicitely and not rely to automatic assignment? And if so how? How do people treat this partial Se-Met issue ? We solved the structure with SeSAD ... if I out MSE I get pretty large negative density peaks on the Se ... if I put MET I get positive peaks ;-) And there are good reasons to believe in partial Se- substitution in this case. Thanks in advance, Tassos

Hi, I'm not sure I see what happens... Could you please send me the PDB file and the command you run. You don't need to send me the data - I can always simulate it. Normally, to do what you want you don't need to provide any selections. Atoms in alternate conformations are detected automatically based on altloc identifiers in PDB, the corresponding constrains are built automatically and the constrained occupancy refinement happens by default. Thanks! Pavel. On 11/4/2008 3:46 AM, Anastassis Perrakis wrote:
Hi there -
I was pleased to read in the manual:
* if no selections are provided (refine.occupancies.individual=None and refine.occupancies.group=None) then phenix.refine by default will refine individual occupancies for atoms that have partial occupancy values in input PDB file (except zeros) and it will perform group constrained occupancy refinement for all atoms in alternative conformations. Occupancies of all other atoms will be fixed. * If selections are provided (refine.occupancies.individual=atom_selection or refine.occupancies.group=atom_selection) then the selected atoms will be added to the list of atoms defined above (atoms with partial occupancies or those in alternative conformations). See this document for examples of atom_selection syntax.
However, I want to refine my MET residues in double conformations, one as MET (AMET) and the other one as MSE (BMSE) with each 0.5 occupancy to start with. When I try that I get:
== ----------Initial model statistics (before refinement)---------- ... |-Occupancies statistics------------------------------------------------------| Sorry: There are atoms with negative occupancies. Check input PDB file.
===
The PDB file has 0.5 and 1.0 occupancies only.
I am also using:
==
refinement.refine.strategy=tls+individual_sites+individual_adp+group_anomalous refinement.refine.adp.tls="chain A" refinement.refine.adp.tls="chain B" refinement.main.number_of_macro_cycles=4 refinement.simulated_annealing.start_temperature=5000 refinement.refine.adp.individual.anisotropic =" chain C" refinement.input.xray_data.labels=I_Mystery(+),SIGI_Mystery(+),I_Mystery(-),SIGI_Mystery(-),merged refinement.refine.anomalous_scatterers { group { selection = "name ZN " f_prime = -0.3 f_double_prime = 2.5 refine = *f_prime *f_double_prime } group { selection = "name SE " f_prime = -5.2 f_double_prime = 3.8 refine = *f_prime *f_double_prime } }
==
Any hints on how to proceed ? Should I define groups for the occupancy refinement explicitely and not rely to automatic assignment? And if so how? How do people treat this partial Se-Met issue ? We solved the structure with SeSAD ... if I out MSE I get pretty large negative density peaks on the Se ... if I put MET I get positive peaks ;-) And there are good reasons to believe in partial Se-substitution in this case.
Thanks in advance,
Tassos ------------------------------------------------------------------------
_______________________________________________ phenixbb mailing list [email protected] http://www.phenix-online.org/mailman/listinfo/phenixbb
|-Occupancies statistics------------------------------------------------------| Sorry: There are atoms with negative occupancies. Check input PDB file.
If it says this, that means there must be an atom or atoms with q<0 in your input PDB file.
refinement.refine.strategy=tls+individual_sites+individual_adp+group_anomalous
Note, this will NOT refine occupancies. To refine occupancies, you need to add "+occupancies" to the line above.
Any hints on how to proceed ?
Just add "+occupancies" and no need to give any selections. All the best! Pavel.

Hi Tassos,
However, I want to refine my MET residues in double conformations, one as MET (AMET) and the other one as MSE (BMSE) with each 0.5 occupancy to start with.
In addition to what Pavel wrote: I just tried out the AMET BMSE mix without giving any manual atom selections, and it works fine for me. (P.S.: I'm thrilled to see this use of the multiple-conformers-with-mixed-residue-names feature, since it was a lot of work.) Ralf

Hi Ralf, for future reference: Could you guys advertise these kind of nuggets a bit more, please? Its great to have this feature in there, but what use is it when only a few people know about it. Did I miss the advert somehow or should I RTFM? Cheers, Carsten -----Original Message----- From: [email protected] [mailto:[email protected]]On Behalf Of Ralf W. Grosse-Kunstleve Sent: Tuesday, November 04, 2008 12:36 PM To: [email protected] Subject: Re: [phenixbb] refine occupancies for MET/MSE double conformations Hi Tassos,
However, I want to refine my MET residues in double conformations, one as MET (AMET) and the other one as MSE (BMSE) with each 0.5 occupancy to start with.
In addition to what Pavel wrote: I just tried out the AMET BMSE mix without giving any manual atom selections, and it works fine for me. (P.S.: I'm thrilled to see this use of the multiple-conformers-with-mixed-residue-names feature, since it was a lot of work.) Ralf _______________________________________________ phenixbb mailing list [email protected] http://www.phenix-online.org/mailman/listinfo/phenixbb
Hi Carsten, I completely agree: every time we have something new, we need to write about it to phenixbb. Even though it might be in the list of new features that comes with the release, repeating is not bad. Pavel. On 11/4/2008 9:53 AM, Schubert, Carsten [PRDUS] wrote:
Hi Ralf,
for future reference: Could you guys advertise these kind of nuggets a bit more, please? Its great to have this feature in there, but what use is it when only a few people know about it. Did I miss the advert somehow or should I RTFM?
Cheers,
Carsten
-----Original Message----- From: [email protected] [mailto:[email protected]]On Behalf Of Ralf W. Grosse-Kunstleve Sent: Tuesday, November 04, 2008 12:36 PM To: [email protected] Subject: Re: [phenixbb] refine occupancies for MET/MSE double conformations
Hi Tassos,
However, I want to refine my MET residues in double conformations, one as MET (AMET) and the other one as MSE (BMSE) with each 0.5 occupancy to start with.
In addition to what Pavel wrote: I just tried out the AMET BMSE mix without giving any manual atom selections, and it works fine for me. (P.S.: I'm thrilled to see this use of the multiple-conformers-with-mixed-residue-names feature, since it was a lot of work.)
Ralf _______________________________________________ phenixbb mailing list [email protected] http://www.phenix-online.org/mailman/listinfo/phenixbb
_______________________________________________ phenixbb mailing list [email protected] http://www.phenix-online.org/mailman/listinfo/phenixbb
for future reference: Could you guys advertise these kind of nuggets a bit more, please? Its great to have this feature in there, but what use is it when only a few people know about it.
Until I saw Tasso's email I thought myself it is a pretty esoteric feature. There is some information here: http://cci.lbl.gov/~rwgk/ccp4nl_2008_08/news.html (I wrote this in Aug; the newsletter is unfortunately seriously delayed). Ralf

I did not know about these features - I just always wanted to do this and in a fit of boredom I tried it since RTPHM suggested it would be supported. I must say that it was a pleasant surprise to find crystallographic software beating my perverse crystallographic imagination ;-) We have an 1.6 A dataset with anomalous signal, Se-Met, two different metals in it (one is Zn ... I think), its a complex between two proteins with each protein having good reasons for differential Se-Met content. That would be so exciting if only it was not two E.coli metal binders that ^&@$%&^$@!^#$^&co-purified (2-3%) with our protein of interest - in the SeMet prep. I am taking my revenge to them subjecting them to weird refinement. Acta F here we come! In the meantime we need to solve the real structure in a pseudo F-centered SG (C2221 according to the holy triage that is) with 3.4 A data. Hurrah - but its more fun. A. On 4 Nov 2008, at 18:53, Schubert, Carsten [PRDUS] wrote:
Hi Ralf,
for future reference: Could you guys advertise these kind of nuggets a bit more, please? Its great to have this feature in there, but what use is it when only a few people know about it. Did I miss the advert somehow or should I RTFM?
Cheers,
Carsten
-----Original Message----- From: [email protected] [mailto:[email protected]]On Behalf Of Ralf W. Grosse-Kunstleve Sent: Tuesday, November 04, 2008 12:36 PM To: [email protected] Subject: Re: [phenixbb] refine occupancies for MET/MSE double conformations
Hi Tassos,
However, I want to refine my MET residues in double conformations, one as MET (AMET) and the other one as MSE (BMSE) with each 0.5 occupancy to start with.
In addition to what Pavel wrote: I just tried out the AMET BMSE mix without giving any manual atom selections, and it works fine for me. (P.S.: I'm thrilled to see this use of the multiple-conformers-with-mixed-residue-names feature, since it was a lot of work.)
Ralf _______________________________________________ phenixbb mailing list [email protected] http://www.phenix-online.org/mailman/listinfo/phenixbb
_______________________________________________ phenixbb mailing list [email protected] http://www.phenix-online.org/mailman/listinfo/phenixbb
Ralf, it's not that uncommon. See for example: http://www.rcsb.org/pdb/files/1ejg.pdb Also, you may want to couple partially occupied residue's side chain with partially occupied water. Pavel. On 11/4/2008 9:36 AM, Ralf W. Grosse-Kunstleve wrote:
Hi Tassos,
However, I want to refine my MET residues in double conformations, one as MET (AMET) and the other one as MSE (BMSE) with each 0.5 occupancy to start with.
In addition to what Pavel wrote: I just tried out the AMET BMSE mix without giving any manual atom selections, and it works fine for me. (P.S.: I'm thrilled to see this use of the multiple-conformers-with-mixed-residue-names feature, since it was a lot of work.)
Ralf _______________________________________________ phenixbb mailing list [email protected] http://www.phenix-online.org/mailman/listinfo/phenixbb

How can I give occupancy 0.5 to my ligands and avoid occupancy refinement? Because the refinement gave me some atoms with occupancy 0.0 and adjacent atoms with occupancy 1.0 in the same ligand. ----- Original Message ----- From: Pavel Afonine To: Ralf W. Grosse-Kunstleve ; PHENIX user mailing list Sent: Tuesday, November 04, 2008 12:15 PM Subject: Re: [phenixbb] refine occupancies for MET/MSE double conformations Ralf, it's not that uncommon. See for example: http://www.rcsb.org/pdb/files/1ejg.pdb Also, you may want to couple partially occupied residue's side chain with partially occupied water. Pavel. On 11/4/2008 9:36 AM, Ralf W. Grosse-Kunstleve wrote: Hi Tassos, However, I want to refine my MET residues in double conformations, one as MET (AMET) and the other one as MSE (BMSE) with each 0.5 occupancy to start with. In addition to what Pavel wrote: I just tried out the AMET BMSE mix without giving any manual atom selections, and it works fine for me. (P.S.: I'm thrilled to see this use of the multiple-conformers-with-mixed-residue-names feature, since it was a lot of work.) Ralf _______________________________________________ phenixbb mailing list [email protected] http://www.phenix-online.org/mailman/listinfo/phenixbb ------------------------------------------------------------------------------ _______________________________________________ phenixbb mailing list [email protected] http://www.phenix-online.org/mailman/listinfo/phenixbb
Hi Maia, just tun off the occupancy refinement. You can do it by removing the "*" in front of "occupancies", as illustrated below: strategy = *individual_sites rigid_body *individual_adp group_adp *tls \ *occupancies *group_anomalous Pavel. On 11/4/2008 11:26 AM, chern wrote:
How can I give occupancy 0.5 to my ligands and avoid occupancy refinement? Because the refinement gave me some atoms with occupancy 0.0 and adjacent atoms with occupancy 1.0 in the same ligand.
----- Original Message ----- *From:* Pavel Afonine mailto:[email protected] *To:* Ralf W. Grosse-Kunstleve mailto:[email protected] ; PHENIX user mailing list mailto:[email protected] *Sent:* Tuesday, November 04, 2008 12:15 PM *Subject:* Re: [phenixbb] refine occupancies for MET/MSE double conformations
Ralf,
it's not that uncommon. See for example: http://www.rcsb.org/pdb/files/1ejg.pdb
Also, you may want to couple partially occupied residue's side chain with partially occupied water.
Pavel.
On 11/4/2008 9:36 AM, Ralf W. Grosse-Kunstleve wrote:
Hi Tassos,
However, I want to refine my MET residues in double conformations, one as MET (AMET) and the other one as MSE (BMSE) with each 0.5 occupancy to start with.
In addition to what Pavel wrote: I just tried out the AMET BMSE mix without giving any manual atom selections, and it works fine for me. (P.S.: I'm thrilled to see this use of the multiple-conformers-with-mixed-residue-names feature, since it was a lot of work.)
Ralf _______________________________________________ phenixbb mailing list [email protected] http://www.phenix-online.org/mailman/listinfo/phenixbb
------------------------------------------------------------------------ _______________________________________________ phenixbb mailing list [email protected] http://www.phenix-online.org/mailman/listinfo/phenixbb
------------------------------------------------------------------------
_______________________________________________ phenixbb mailing list [email protected] http://www.phenix-online.org/mailman/listinfo/phenixbb
You can also do group occupancy refinement: refine one occupancy for the whole ligand - in this case you don't need to put an ad hoc value. The current limitation (bug) is that the starting occupancy values in the input PDB file for that ligand must be all 1.0. Pavel. On 11/4/2008 11:26 AM, chern wrote:
How can I give occupancy 0.5 to my ligands and avoid occupancy refinement? Because the refinement gave me some atoms with occupancy 0.0 and adjacent atoms with occupancy 1.0 in the same ligand.
----- Original Message ----- *From:* Pavel Afonine mailto:[email protected] *To:* Ralf W. Grosse-Kunstleve mailto:[email protected] ; PHENIX user mailing list mailto:[email protected] *Sent:* Tuesday, November 04, 2008 12:15 PM *Subject:* Re: [phenixbb] refine occupancies for MET/MSE double conformations
Ralf,
it's not that uncommon. See for example: http://www.rcsb.org/pdb/files/1ejg.pdb
Also, you may want to couple partially occupied residue's side chain with partially occupied water.
Pavel.
On 11/4/2008 9:36 AM, Ralf W. Grosse-Kunstleve wrote:
Hi Tassos,
However, I want to refine my MET residues in double conformations, one as MET (AMET) and the other one as MSE (BMSE) with each 0.5 occupancy to start with.
In addition to what Pavel wrote: I just tried out the AMET BMSE mix without giving any manual atom selections, and it works fine for me. (P.S.: I'm thrilled to see this use of the multiple-conformers-with-mixed-residue-names feature, since it was a lot of work.)
Ralf _______________________________________________ phenixbb mailing list [email protected] http://www.phenix-online.org/mailman/listinfo/phenixbb
------------------------------------------------------------------------ _______________________________________________ phenixbb mailing list [email protected] http://www.phenix-online.org/mailman/listinfo/phenixbb
------------------------------------------------------------------------
_______________________________________________ phenixbb mailing list [email protected] http://www.phenix-online.org/mailman/listinfo/phenixbb

Hi Pavel, Actually, I have 2 practical problems (see attachment) which I haven't been able to deal with so far using phenix.refine (latest version). Ligand A shows 2 stacking modes (A1 and A2) above an FMN cofactor. The A2 conformation also overlaps partially with another partially occupied ligand B. How do I couple the occupancy refinements of these interrelated ligands? Until now, I solved it by refining the occupancy as if ligand A has 3 modes; A1, A2 and A3 (= B). Otherwise, ligand B is always pushed out of its density due to the anti-bumping restraints. This is high res data (~1.5 A), so pretty decisive about atomic positions. The same question goes for the coupling of conformation C1 of an Ile residue and water D on one hand, and conformation C2 of that Ile on the other hand. Thus far I've only been able to define these relations in SHELXL, but it's not entirely clear to me how to do this in phenix.refine (which I prefer for this dataset). Many thanks in advance, Jonathan Elegheert PhD Student L-ProBE, Ghent University Belgium Pavel Afonine wrote:
Ralf,
it's not that uncommon. See for example: http://www.rcsb.org/pdb/files/1ejg.pdb
Also, you may want to couple partially occupied residue's side chain with partially occupied water.
Pavel.
On 11/4/2008 9:36 AM, Ralf W. Grosse-Kunstleve wrote:
Hi Tassos,
However, I want to refine my MET residues in double conformations, one as MET (AMET) and the other one as MSE (BMSE) with each 0.5 occupancy to start with.
In addition to what Pavel wrote: I just tried out the AMET BMSE mix without giving any manual atom selections, and it works fine for me. (P.S.: I'm thrilled to see this use of the multiple-conformers-with-mixed-residue-names feature, since it was a lot of work.)
Ralf _______________________________________________ phenixbb mailing list [email protected] http://www.phenix-online.org/mailman/listinfo/phenixbb
------------------------------------------------------------------------
_______________________________________________ phenixbb mailing list [email protected] http://www.phenix-online.org/mailman/listinfo/phenixbb
{kind=link}
{kind=link}
Hi Jonathan, currently it's not possible (if I correctly understand what you are trying to do), but it will be available (hopefully) in the next version. Pavel. On 11/11/2008 3:13 AM, Jonathan Elegheert wrote:
Hi Pavel,
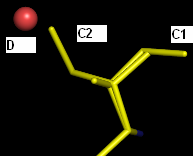
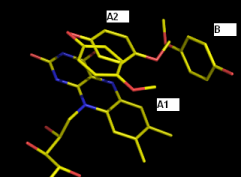
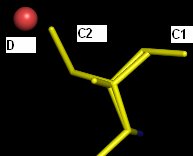
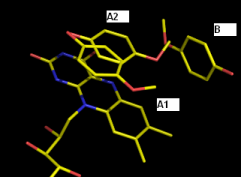
Actually, I have 2 practical problems (see attachment) which I haven't been able to deal with so far using phenix.refine (latest version).
Ligand A shows 2 stacking modes (A1 and A2) above an FMN cofactor. The A2 conformation also overlaps partially with another partially occupied ligand B. How do I couple the occupancy refinements of these interrelated ligands? Until now, I solved it by refining the occupancy as if ligand A has 3 modes; A1, A2 and A3 (= B). Otherwise, ligand B is always pushed out of its density due to the anti-bumping restraints. This is high res data (~1.5 A), so pretty decisive about atomic positions.
The same question goes for the coupling of conformation C1 of an Ile residue and water D on one hand, and conformation C2 of that Ile on the other hand.
Thus far I've only been able to define these relations in SHELXL, but it's not entirely clear to me how to do this in phenix.refine (which I prefer for this dataset).
Many thanks in advance,
Jonathan Elegheert PhD Student L-ProBE, Ghent University Belgium
Pavel Afonine wrote:
Ralf,
it's not that uncommon. See for example: http://www.rcsb.org/pdb/files/1ejg.pdb
Also, you may want to couple partially occupied residue's side chain with partially occupied water.
Pavel.
On 11/4/2008 9:36 AM, Ralf W. Grosse-Kunstleve wrote:
Hi Tassos,
However, I want to refine my MET residues in double conformations, one as MET (AMET) and the other one as MSE (BMSE) with each 0.5 occupancy to start with.
In addition to what Pavel wrote: I just tried out the AMET BMSE mix without giving any manual atom selections, and it works fine for me. (P.S.: I'm thrilled to see this use of the multiple-conformers-with-mixed-residue-names feature, since it was a lot of work.)
Ralf _______________________________________________ phenixbb mailing list [email protected] http://www.phenix-online.org/mailman/listinfo/phenixbb
------------------------------------------------------------------------
_______________________________________________ phenixbb mailing list [email protected] http://www.phenix-online.org/mailman/listinfo/phenixbb
------------------------------------------------------------------------
------------------------------------------------------------------------
------------------------------------------------------------------------
_______________________________________________ phenixbb mailing list [email protected] http://www.phenix-online.org/mailman/listinfo/phenixbb
{kind=link}
{kind=link}
Hi Jonathan, currently it's not possible (if I correctly understand what you are trying to do), but it will be available (hopefully) in the next version. Pavel. On 11/11/2008 3:13 AM, Jonathan Elegheert wrote:
Hi Pavel,
Actually, I have 2 practical problems (see attachment) which I haven't been able to deal with so far using phenix.refine (latest version).
Ligand A shows 2 stacking modes (A1 and A2) above an FMN cofactor. The A2 conformation also overlaps partially with another partially occupied ligand B. How do I couple the occupancy refinements of these interrelated ligands? Until now, I solved it by refining the occupancy as if ligand A has 3 modes; A1, A2 and A3 (= B). Otherwise, ligand B is always pushed out of its density due to the anti-bumping restraints. This is high res data (~1.5 A), so pretty decisive about atomic positions.
The same question goes for the coupling of conformation C1 of an Ile residue and water D on one hand, and conformation C2 of that Ile on the other hand.
Thus far I've only been able to define these relations in SHELXL, but it's not entirely clear to me how to do this in phenix.refine (which I prefer for this dataset).
Many thanks in advance,
Jonathan Elegheert PhD Student L-ProBE, Ghent University Belgium
Pavel Afonine wrote:
Ralf,
it's not that uncommon. See for example: http://www.rcsb.org/pdb/files/1ejg.pdb
Also, you may want to couple partially occupied residue's side chain with partially occupied water.
Pavel.
On 11/4/2008 9:36 AM, Ralf W. Grosse-Kunstleve wrote:
Hi Tassos,
However, I want to refine my MET residues in double conformations, one as MET (AMET) and the other one as MSE (BMSE) with each 0.5 occupancy to start with.
In addition to what Pavel wrote: I just tried out the AMET BMSE mix without giving any manual atom selections, and it works fine for me. (P.S.: I'm thrilled to see this use of the multiple-conformers-with-mixed-residue-names feature, since it was a lot of work.)
Ralf _______________________________________________ phenixbb mailing list [email protected] http://www.phenix-online.org/mailman/listinfo/phenixbb
------------------------------------------------------------------------
_______________________________________________ phenixbb mailing list [email protected] http://www.phenix-online.org/mailman/listinfo/phenixbb
participants (8)
-
 Anastassis Perrakis
Anastassis Perrakis -
 chern
chern -
 Jonathan Elegheert
Jonathan Elegheert -
 Pavel Afonine
Pavel Afonine -
Pavel Afonine
-
 Peter Zwart
Peter Zwart -
 Ralf W. Grosse-Kunstleve
Ralf W. Grosse-Kunstleve -
 Schubert, Carsten [PRDUS]
Schubert, Carsten [PRDUS]